Paired Mass Distance(PMD) analysis for GC/LC-MS based non-targeted analysis
Miao Yu
2026-04-22
Source:vignettes/globalstd.Rmd
globalstd.RmdIntroduction of Paired Mass Distance analysis
pmd package use Paired Mass Distance (PMD) relationship
to analysis the GC/LC-MS based non-targeted data. PMD means the distance
between two masses or mass to charge ratios. In mass spectrometry, PMD
would keep the same value between two masses and two mass to charge
ratios(m/z). There are two kinds of PMD involved in this package: PMD
from the same compound and PMD from different compounds. In GC/LC-MS or
XCMS based non-targeted data analysis, peaks could be separated by
chronograph and same compound means ions from similar retention times or
ions co-eluted by certain column.
PMD from the same compound
For MS1 full scan data, we could build retention time(RT) bins to assign peaks into different RT groups by retention time hierarchical clustering analysis. For each RT group, the peaks should come from same compounds or co-elutes. If certain PMD appeared in multiple RT groups, it would be related to the relationship about adducts, neutral loss, isotopologues or common fragments ions.
PMD from different compounds
The peaks from different retention time groups would like to be different compounds separated by chronograph. The PMD would reflect the relationship about homologous series or chemical reactions.
GlobalStd algorithm use the PMD within same RT group to find independent peaks among certain data set. Then, structure/reaction directed analysis use PMD from different RT groups to screen important compounds or reactions.
Data format
The input data should be a list object with at least two
elements from a peaks list:
- mass to charge ratio with name of
mz, high resolution mass spectrometry is required - retention time with name of
rt
However, I suggested to add intensity and group information to the list for validation of PMD analysis.
In this package, a data set from in vivo solid phase micro-extraction(SPME) was attached. This data set contain 9 samples from 3 fish with triplicates samples for each fish. Here is the data structure:
library(pmd)
data("spmeinvivo")
str(spmeinvivo)
#> List of 4
#> $ data : num [1:1459, 1:9] 1095 10439 10154 2797 90211 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:1459] "100.1/170" "100.5/86" "101/85" "103.1/348" ...
#> .. ..$ : chr [1:9] "1405_Fish1_F1" "1405_Fish1_F2" "1405_Fish1_F3" "1405_Fish2_F1" ...
#> $ group:'data.frame': 9 obs. of 2 variables:
#> ..$ sample_name : chr [1:9] "1405_Fish1_F1" "1405_Fish1_F2" "1405_Fish1_F3" "1405_Fish2_F1" ...
#> ..$ sample_group: chr [1:9] "fish1" "fish1" "fish1" "fish2" ...
#> $ mz : num [1:1459] 100 101 101 103 104 ...
#> $ rt : num [1:1459] 170.2 86.3 84.9 348.1 48.8 ...You could build this list or mzrt object
from the xcms objects via enviGCMS package.
When you have a xcmsSet object or XCMSnExp
object named xset, you could use
enviGCMS::getmzrt(xset) to get such list. Of course you
could build such list by yourself.
GlobalStd algorithm
GlobalStd algorithm try to find independent peaks among certain peaks list. The first step is retention time hierarchical clustering analysis. The second step is to find the relationship among adducts, neutral loss, isotopologues and common fragments ions. The third step is to screen the independent peaks.
Here is a workflow for this algorithm:
knitr::include_graphics('https://yufree.github.io/presentation/figure/GlobalStd.png')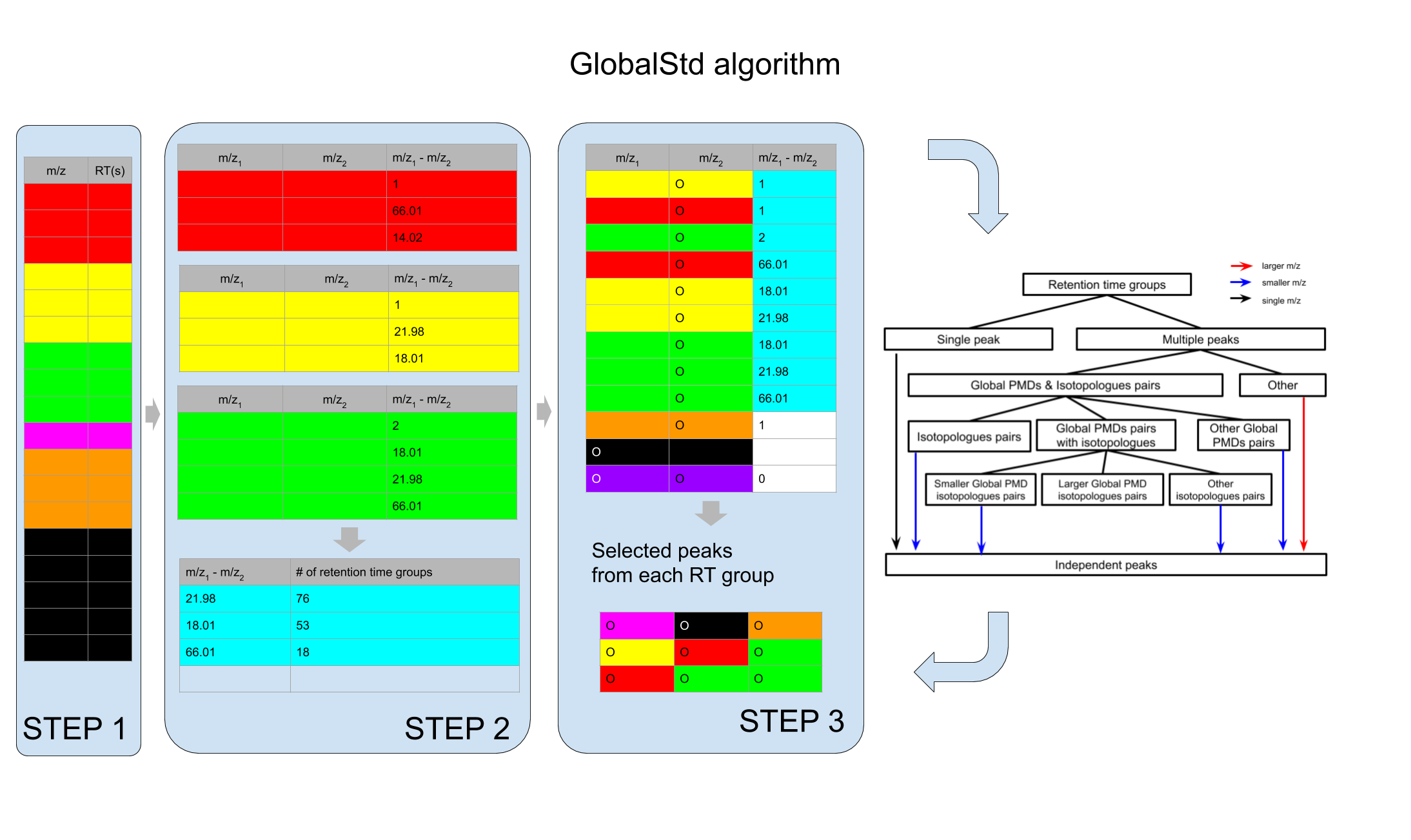
STEP1: Retention time hierarchical clustering
pmd <- getpaired(spmeinvivo)
#> 75 retention time clusters found.
#> Using ng = 15
#> 5 unique PMDs retained.
#> The unique within RT clusters high frequency PMD(s) is(are) 28.03 21.98 44.03 17.03 18.01.
#> 409 isotope peaks found.
#> 109 multiple charged isotope peaks found.
#> 4 multiple charged peaks found.
#> 346 paired peaks found.
plotrtg(pmd)
This plot would show the distribution of RT groups. The
rtcutoff in function getpaired could be used
to set the cutoff of the distances in retention time hierarchical
clustering analysis. Retention time cluster cutoff should fit the peak
picking algorithm. For HPLC, 10 is suggested and 5 could be used for
UPLC.
Global PMD’s retention time group numbers should be around 20 percent
of the retention time cluster numbers. For example, if you find 100
retention time clusters, I suggested you use 20 as the cutoff of
empirical global PMD’s retention time group numbers. If you don’t
specifically assign a value to ng, the algorithm will
select such recommendation by default setting.
Take care of the retention time cluster with lots of peaks. In this case, such cluster could be co-eluted compounds on certain column. It would be wise to trim the retention time window for high quality peaks. Another important hint is that pre-filter your peak list by black samples or other quality control samples. Otherwise the running time would be long and lots of pmd relationship would be just from noise.
STEP2: Relationship among adducts, neutral loss, isotopologues and common fragments ions
The ng in function getpaired could be used
to set cutoff of global PMD’s retention time group numbers. If
ng is 10, at least 10 of the retention time groups should
contain the shown PMD relationship. You could use
plotpaired to show the distribution.
plotpaired(pmd)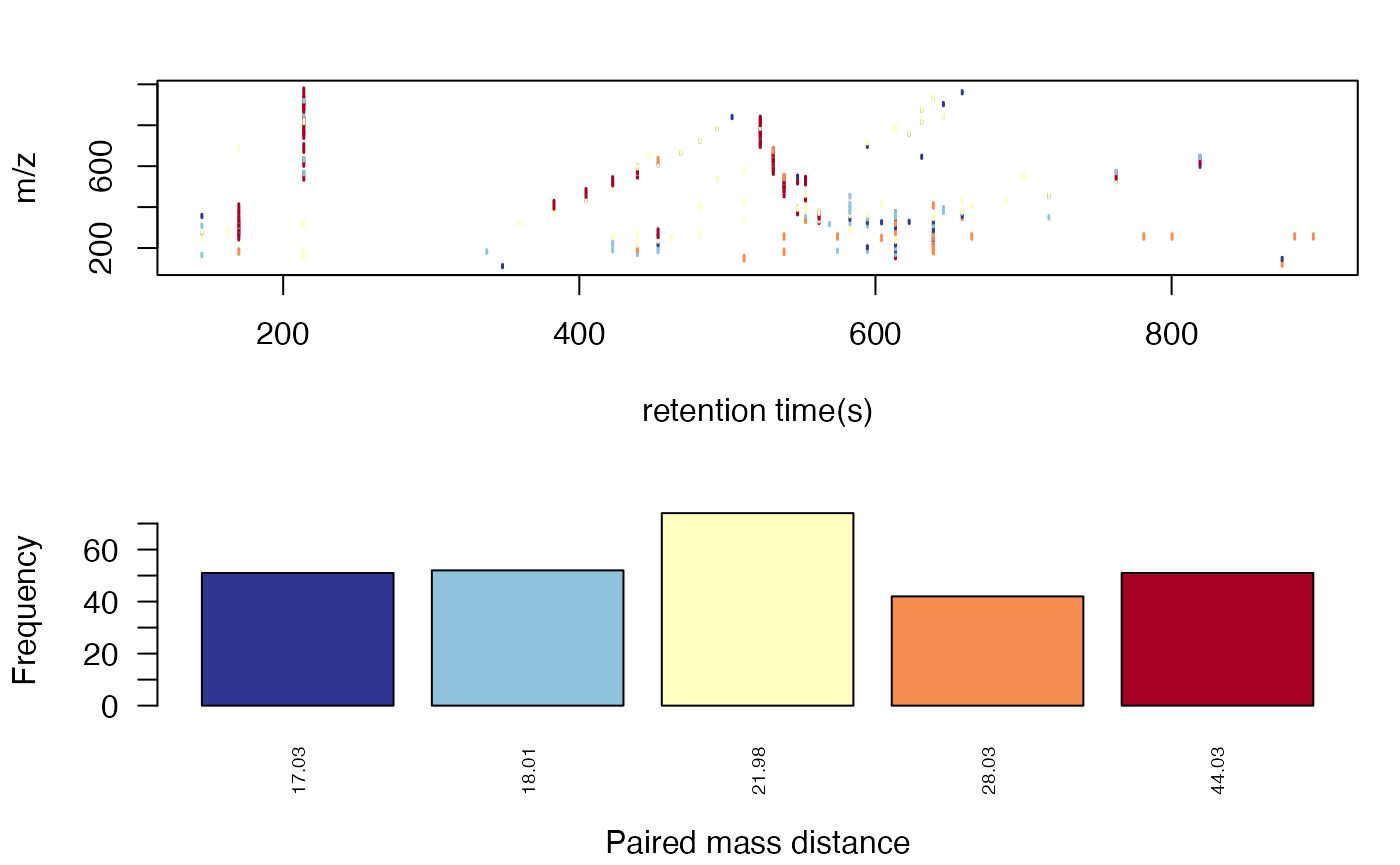
You could also show the distribution of PMD relationship by index:
# show the unique PMD found by getpaired function
for(i in 1:length(unique(round(pmd$paired$diff,2)))){
diff <- unique(round(pmd$paired$diff,2))[i]
index <- round(pmd$paired$diff,2)== diff
plotpaired(pmd,index)
}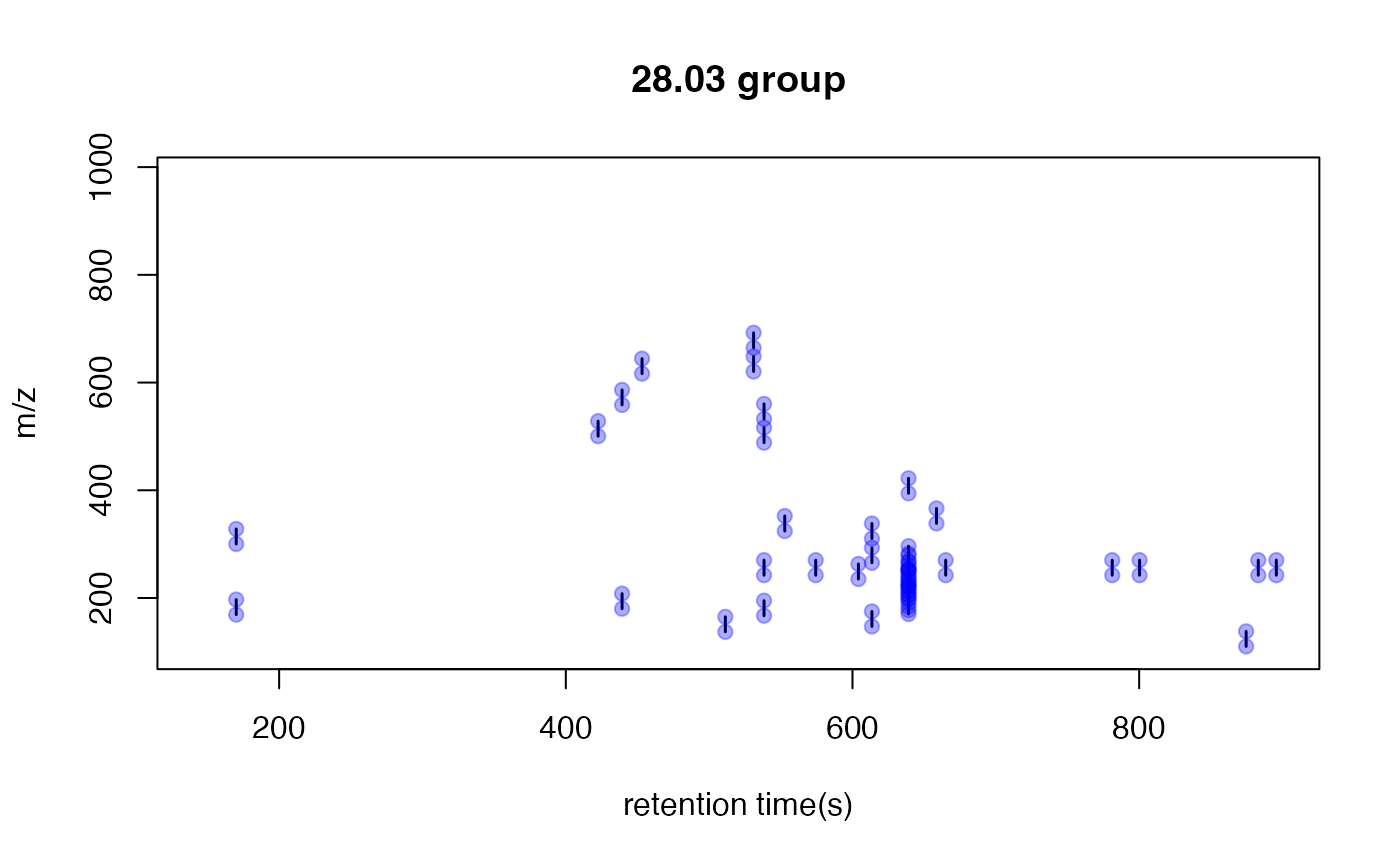
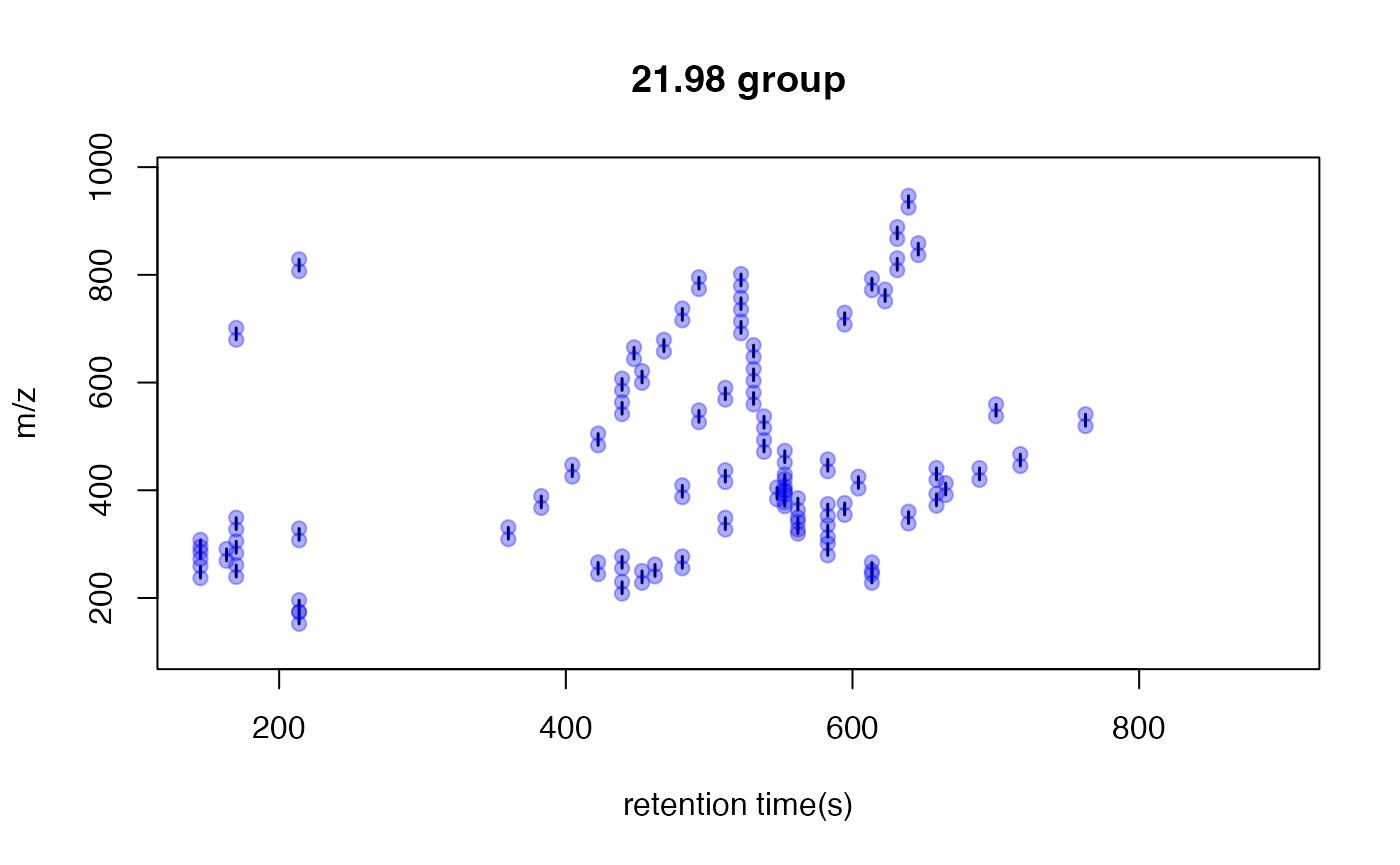
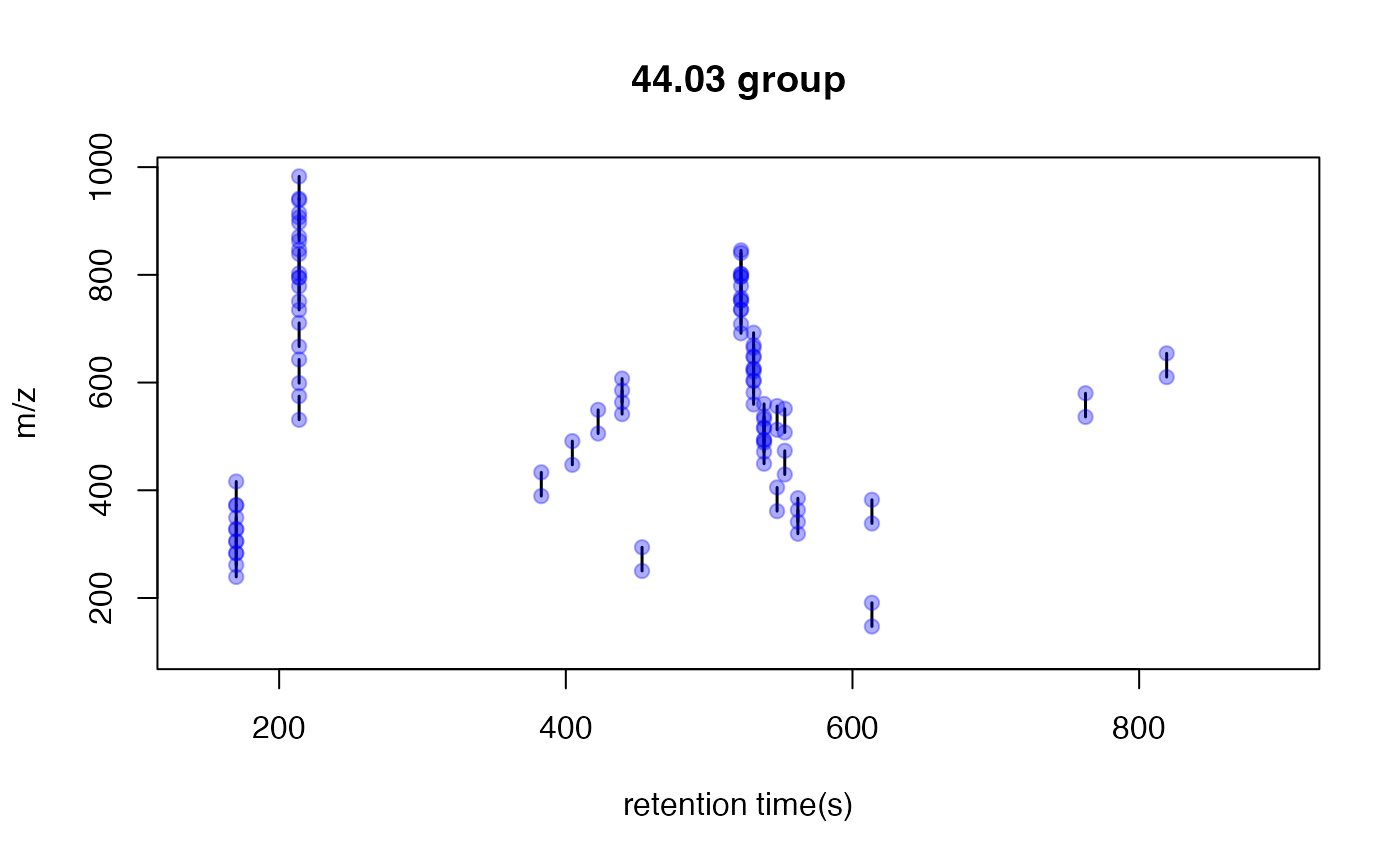


This is an easy way to find potential adducts of the data by high frequency PMD from the same compound. For example, 21.98 Da could be the mass distances between and . In this case, user could find the potential adducts or neutral loss even when they have no preferred adducts list. If one adduct exist in certain analytical system, the high frequency PMD will reveal such relationship. The high frequency PMD list could also be used to check the fragmental pattern of in-source reactions as long as such patterns are popular among all collected ions.
STEP3: Screen the independent peaks
You could use getstd function to get the independent
peaks. Independent peaks mean the peaks list removing the redundant
peaks such as adducts, neutral loss, isotopologues and comment fragments
ions found by PMD analysis in STEP2. Ideally, those peaks could be
molecular ions while they might still contain redundant peaks.
std <- getstd(pmd)
#> 8 group(s) have single peaks 14 23 32 33 54 55 56 75
#> 11 group(s) with multiple peaks while no isotope/paired relationship 4 5 7 8 11 ... 42 49 68 72 73
#> 9 group(s) with isotope without paired relationship 2 9 22 26 52 62 64 66 70
#> 4 group(s) with paired without isotope relationship 1 10 15 18
#> 43 group(s) with both paired and isotope relationship 3 6 12 13 16 ... 65 67 69 71 74
#> 291 standard masses identified.Here you could plot the peaks by plotstd function to
show the distribution of independent peaks:
plotstd(std)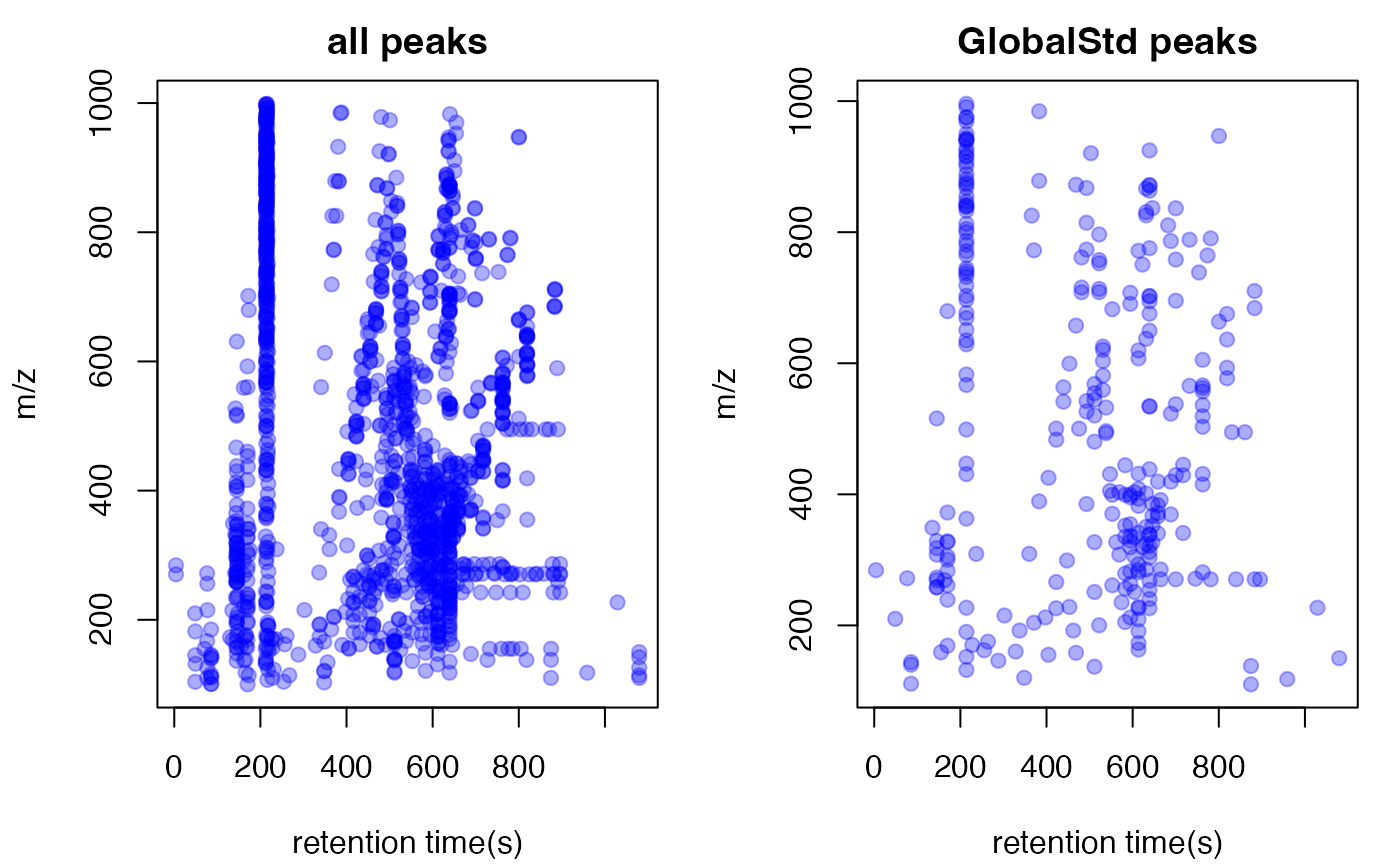
You could also plot the peaks distribution by assign a retention time
group via plotstdrt:
par(mfrow = c(2,3))
plotstdrt(std,rtcluster = 23,main = 'Retention time group 23')
plotstdrt(std,rtcluster = 9,main = 'Retention time group 9')
plotstdrt(std,rtcluster = 18,main = 'Retention time group 18')
plotstdrt(std,rtcluster = 67,main = 'Retention time group 67')
plotstdrt(std,rtcluster = 49,main = 'Retention time group 49')
plotstdrt(std,rtcluster = 6,main = 'Retention time group 6')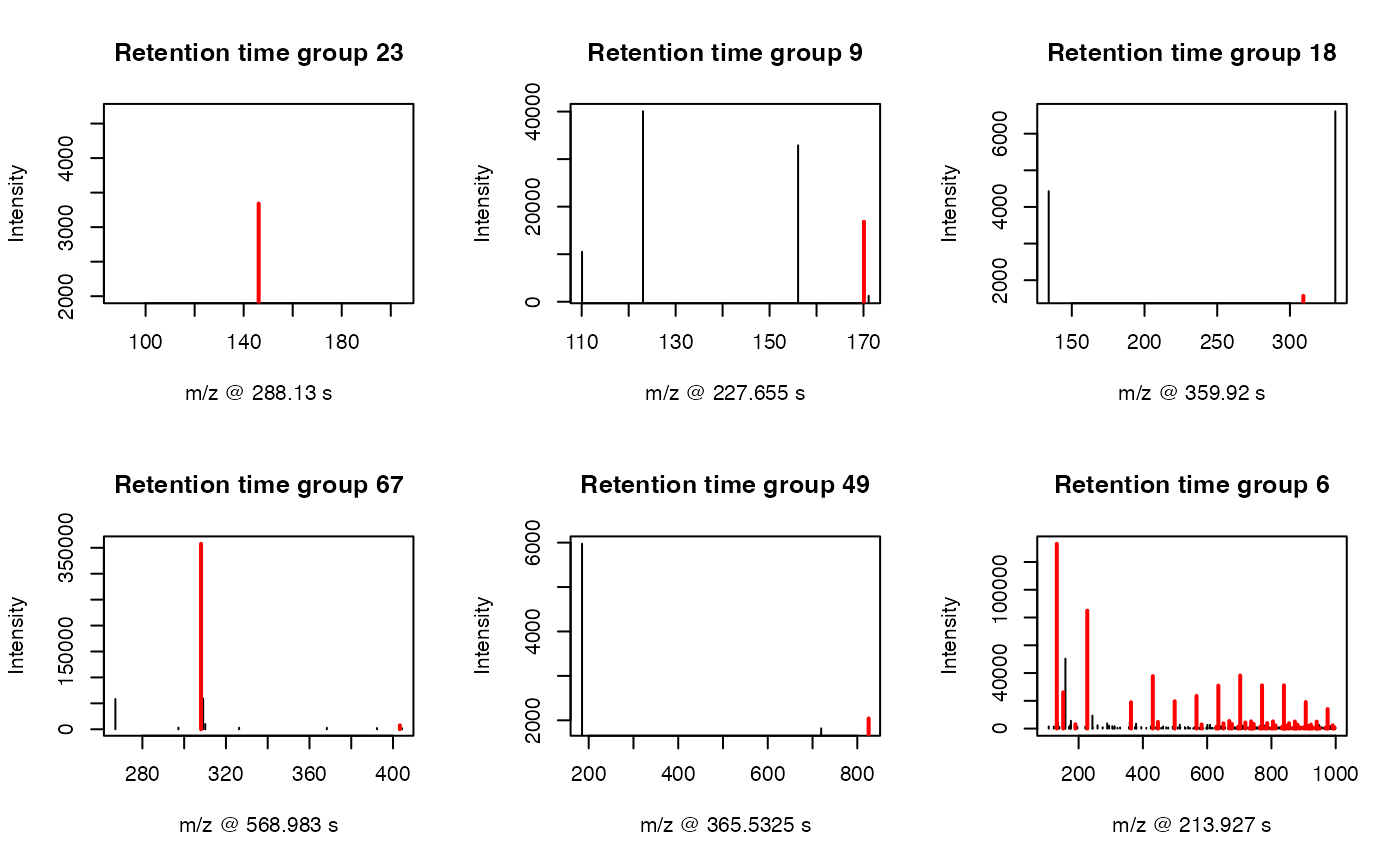
Extra filter with correlation coefficient cutoff
Original GlobalStd algorithm only use mass to charge ratio and retention time of peaks to select independent peaks. However, if intensity data across samples are available, correlation coefficient of paired ions could be used to further filter the random noise in high frequency PMDs. You could set up cutoff of Pearson Correlation Coefficient between peaks to refine the peaks selected by GlobalStd within same retention time groups. In this case, the numbers of selected independent peaks will be further reduced. When you use this parameter, make sure the intensity data are from real samples instead of blank samples, which will affect the calculation of correlation coefficient.
std2 <- globalstd(pmd,corcutoff = 0.9)
#> 75 retention time clusters found.
#> Using ng = 15
#> 2 unique PMDs retained.
#> The unique within RT clusters high frequency PMD(s) is(are) 21.98 17.03.
#> 242 isotope peaks found.
#> 63 multiple charged isotope peaks found.
#> 3 multiple charged peaks found.
#> 120 paired peaks found.
#> 8 group(s) have single peaks 14 23 32 33 54 55 56 75
#> 23 group(s) with multiple peaks while no isotope/paired relationship 2 4 5 7 8 ... 68 69 70 72 73
#> 17 group(s) with isotope without paired relationship 9 12 20 22 24 ... 60 64 66 67 71
#> 3 group(s) with paired without isotope relationship 1 53 74
#> 24 group(s) with both paired and isotope relationship 3 6 13 16 17 ... 48 58 61 63 65
#> 206 standard masses identified.Validation by principal components analysis(PCA)
You need to check the GlobalStd algorithm’s results by principal components analysis(PCA). If we removed too much peaks containing information, the score plot of reduced data set would show great changes.
library(enviGCMS)
par(mfrow = c(2,2),mar = c(4,4,2,1)+0.1)
plotpca(std$data,lv = as.numeric(as.factor(std$group$sample_group)),main = "all peaks")
plotpca(std$data[std$stdmassindex,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(std$stdmassindex),"independent peaks"))
plotpca(std2$data[std2$stdmassindex,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(std2$stdmassindex),"reduced independent peaks"))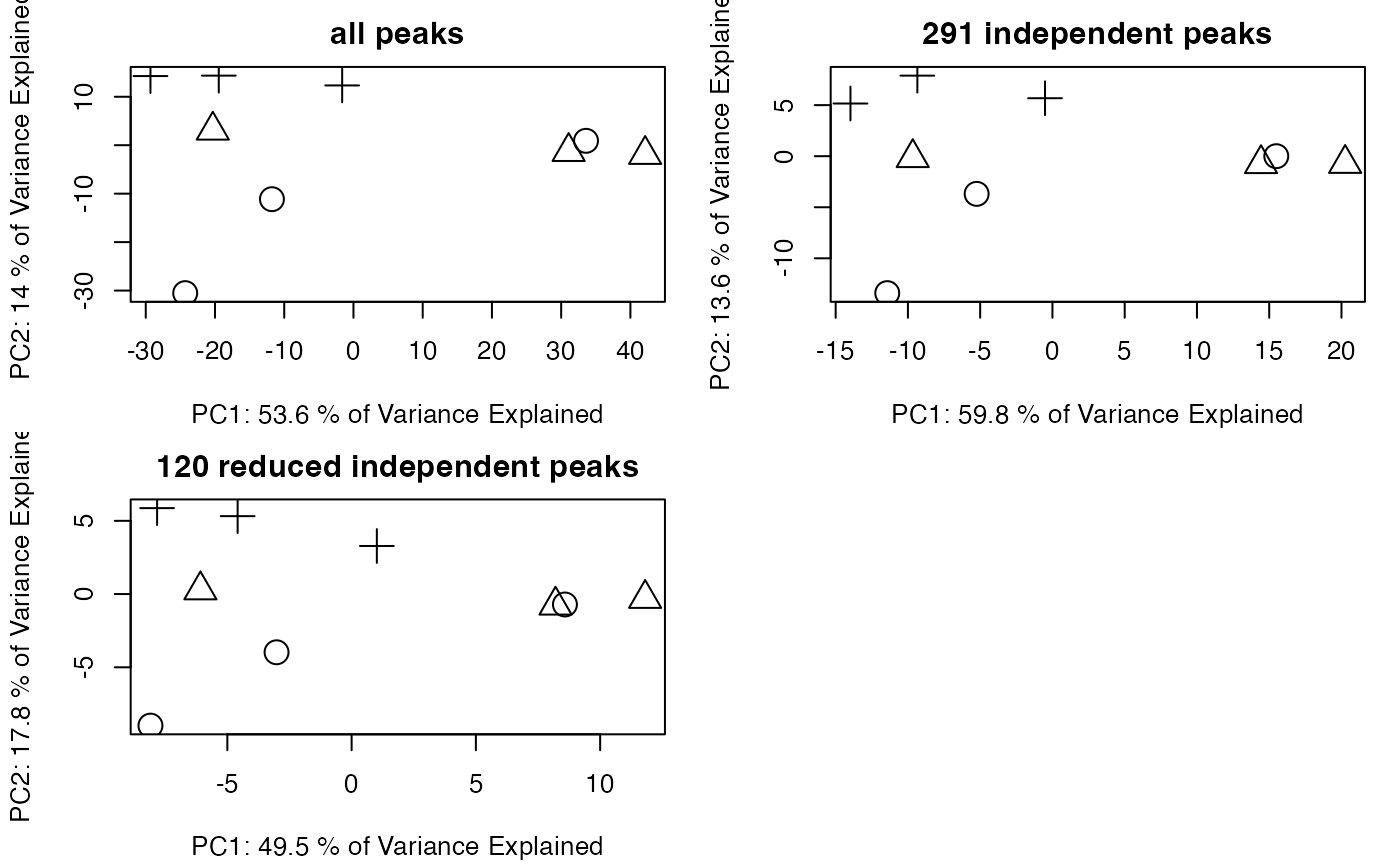
You might find original GlobalStd algorithm show a similar PCA score plot with original data while GlobalStd algorithm considering intensity data seems change the profile. The major reason is that correlation coefficient option in the algorithm will remove the paired ions without strong correlation. It will be aggressive to remove low intensity peaks, which are vulnerable by baseline noise. However, such options would be helpful if you only concern high quality peaks for following analysis. Otherwise, original GlobalStd will keep the most information for discover purpose.
Comparison with other pseudo spectra extraction method
GlobalStd algorithm in pmd package could be treated as a
method to extract pseudo spectra. You could use
getpseudospectrum to get peaks groups information for all
GlobalStd peaks. This function would consider the merge of GlobalStd
peaks when certain peak is involved in multiple clusters. Then you could
choose export peaks with the highest intensities or base peaks in each
GlobalStd merged peaks groups. Meanwhile, you could also include the
correlation coefficient cutoff to further improve the data quality.
stdcluster <- getpseudospectrum(std)
#> 75 retention time clusters found.
#> Using ng = 15
#> 5 unique PMDs retained.
#> The unique within RT clusters high frequency PMD(s) is(are) 28.03 21.98 44.03 17.03 18.01.
#> 409 isotope peaks found.
#> 109 multiple charged isotope peaks found.
#> 4 multiple charged peaks found.
#> 346 paired peaks found.
#> 245 pseudo spectrum found.
#> 0.84 peaks covered by PMD relationship.
# extract the first pseudospectra for retention time cluster 37
idx <- stdcluster$pseudo$sid=='37@1'
plot(stdcluster$pseudo$mz[idx],stdcluster$pseudo$ins[idx],type = 'h',xlab = 'm/z',ylab = 'intensity',main = 'pseudo spectra for retention time cluster 37')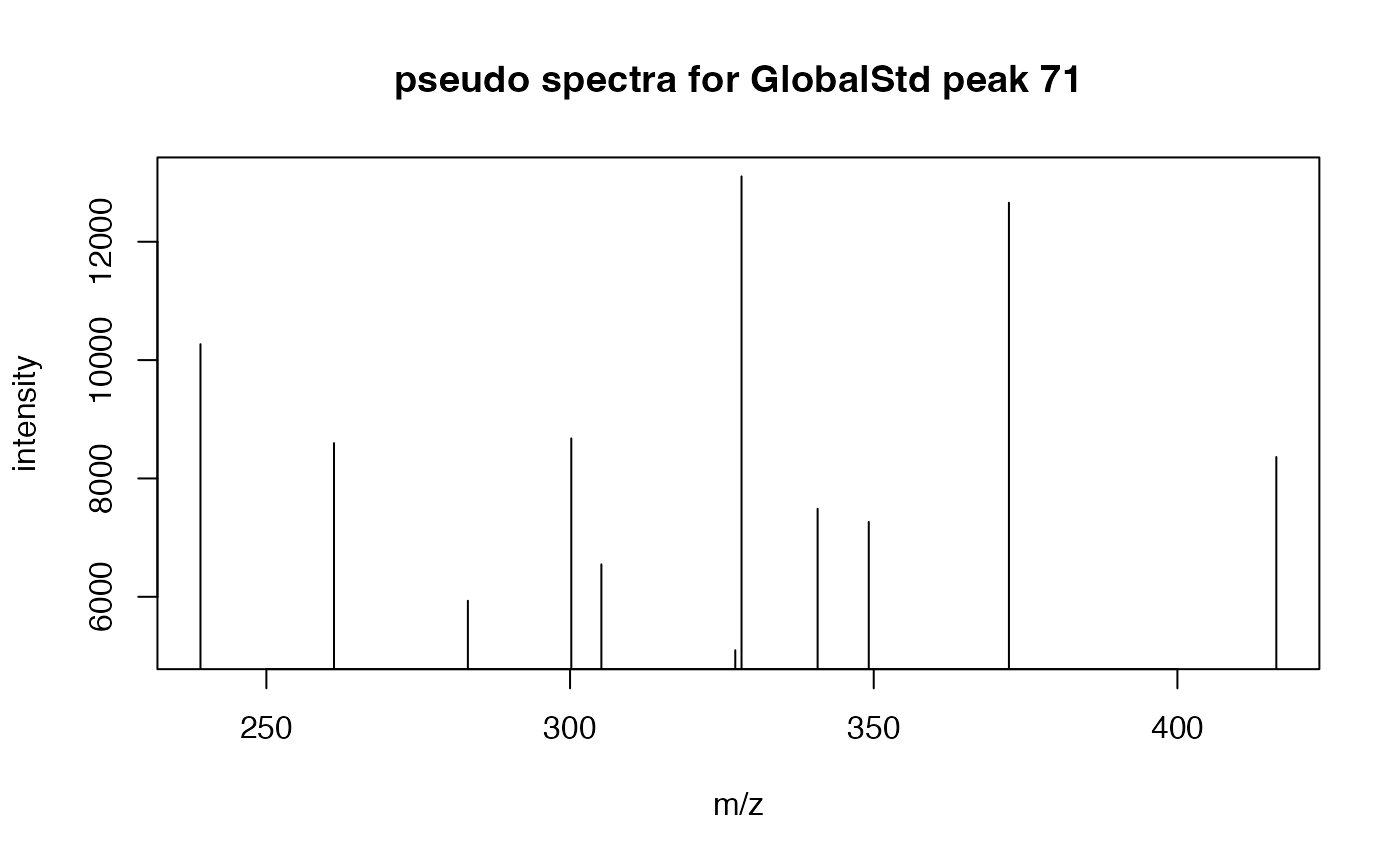
# considering the correlation coefficient cutoff
stdcluster2 <- getpseudospectrum(std, corcutoff = 0.9)
#> 75 retention time clusters found.
#> Using ng = 15
#> 2 unique PMDs retained.
#> The unique within RT clusters high frequency PMD(s) is(are) 21.98 17.03.
#> 242 isotope peaks found.
#> 63 multiple charged isotope peaks found.
#> 3 multiple charged peaks found.
#> 120 paired peaks found.
#> 80 pseudo spectrum found.
#> 0.97 peaks covered by PMD relationship.We supplied getcorpseudospectrum to find peaks groups by
correlation analysis only. The base peaks of correlation cluster were
selected to stand for the compounds.
corcluster <- getcorpseudospectrum(spmeinvivo)
#> 75 retention time cluster found.
#> 141 pseudo spectrum found.
#> 0.72 peaks covered by PMD relationship.
# extract pseudospectra 37@1
peak <- corcluster$pseudo[corcluster$pseudo$sid == '37@1',]
plot(peak$ins~peak$mz,type = 'h',xlab = 'm/z',ylab = 'intensity',main = 'pseudo spectra for correlation cluster')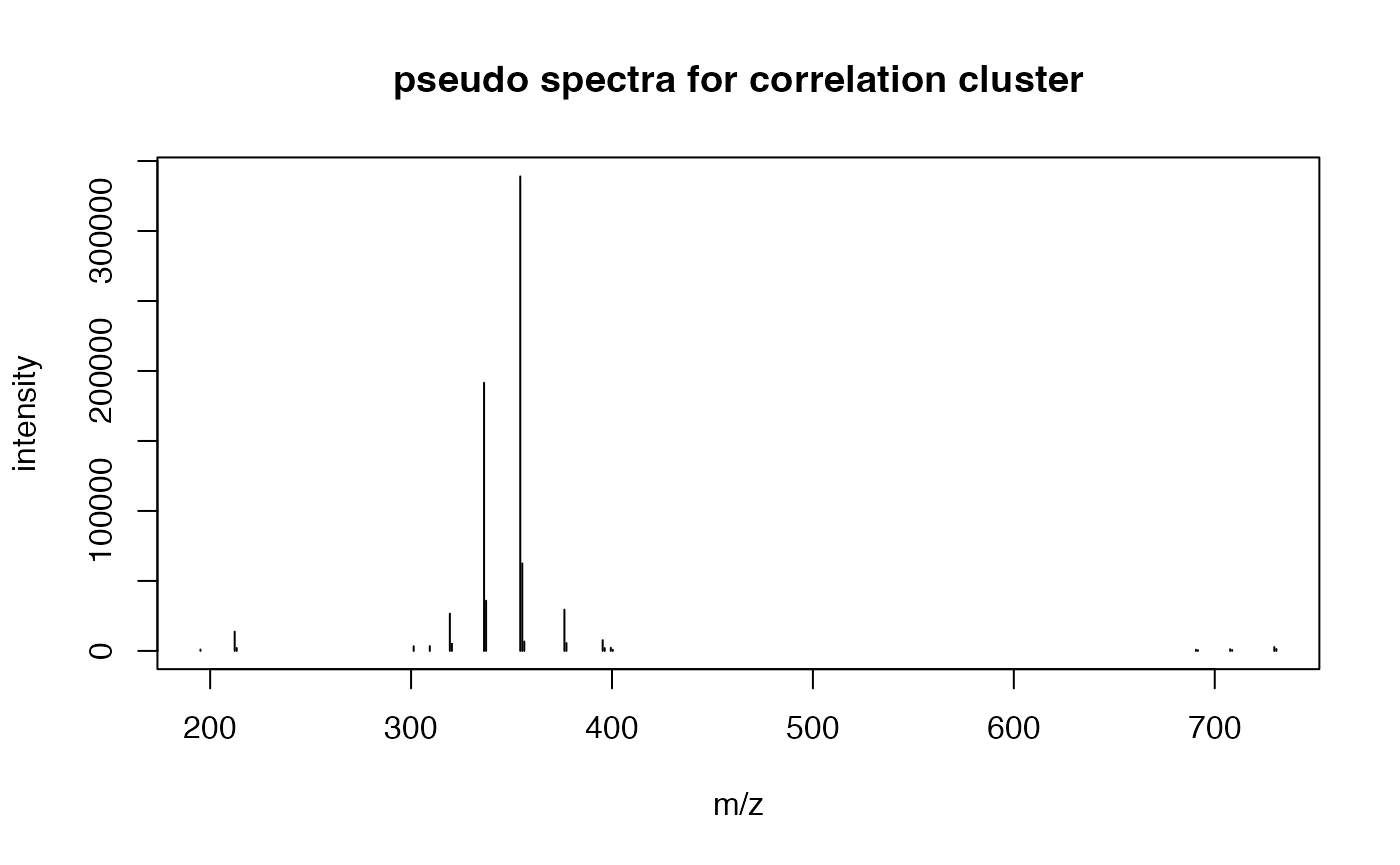
Then we could compare the reduced result using multiple similarity
factors. A good peak selection algorithm should maintain the structural
consistency (e.g. Normalized Spectral Entropy, RV Coefficient, and
Mantel test). We supplied getsim function to output PCASF,
RV, Mantel correlation, and Spectral Entropy ratio between the original
and reduced datasets.
par(mfrow = c(2,3),mar = c(4,4,2,1)+0.1)
plotpca(std$data[std$stdmassindex,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(std$stdmassindex),"independent peaks"))
plotpca(std$data[stdcluster$stdmassindex2,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(stdcluster$stdmassindex2),"independent base peaks"))
plotpca(std$data[stdcluster2$stdmassindex2,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(stdcluster2$stdmassindex2),"independent reduced base peaks"))
plotpca(std$data[corcluster$stdmassindex,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(corcluster$stdmassindex),"peaks without correlationship"))
plotpca(std$data[corcluster$stdmassindex2,],lv = as.numeric(as.factor(std$group$sample_group)),main = paste(sum(corcluster$stdmassindex2),"base peaks without correlationship"))
plotpca(std$data,lv = as.numeric(as.factor(std$group$sample_group)),main = paste(nrow(std$data),"all peaks"))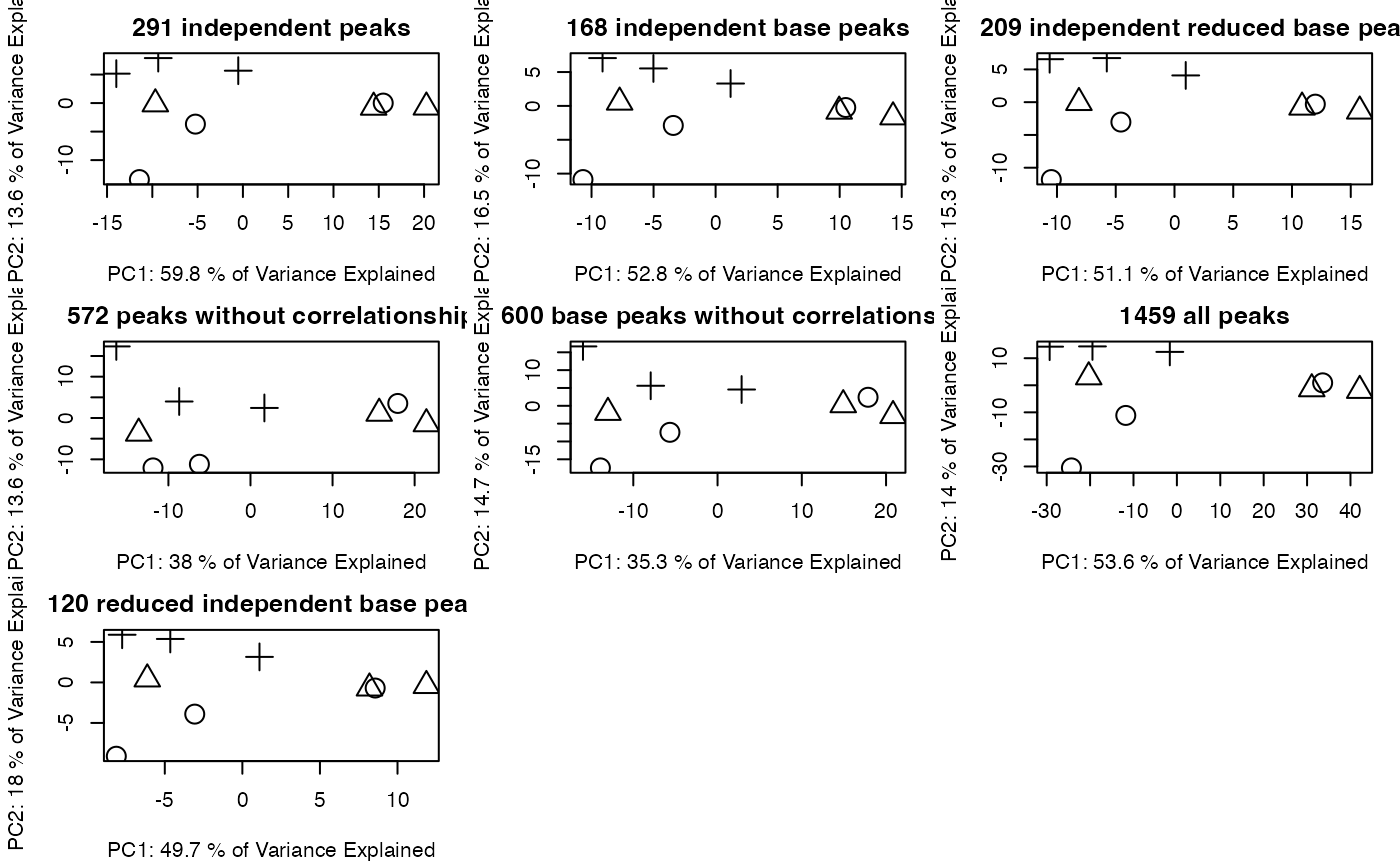
# Evaluate multiple algorithms against original data
getsim(std$data, std$data[std$stdmassindex,])
#> PCASF RV Mantel_Pearson Mantel_Spearman
#> NA 0.9744418 0.9680196 0.9554698
#> Spectral_Entropy_x Spectral_Entropy_y Entropy_Ratio NSE_x
#> 0.8000474 0.6874659 0.8592815 0.3641172
#> NSE_y
#> 0.3128792
getsim(std$data, std$data[stdcluster$stdmassindex2,])
#> PCASF RV Mantel_Pearson Mantel_Spearman
#> NA 0.8599917 0.8371042 0.8404118
#> Spectral_Entropy_x Spectral_Entropy_y Entropy_Ratio NSE_x
#> 0.8000474 0.7130890 0.8913086 0.3641172
#> NSE_y
#> 0.3245408
getsim(std$data, std$data[corcluster$stdmassindex2,])
#> PCASF RV Mantel_Pearson Mantel_Spearman
#> NA 0.7673804 0.7300709 0.7122265
#> Spectral_Entropy_x Spectral_Entropy_y Entropy_Ratio NSE_x
#> 0.8000474 0.4959912 0.6199523 0.3641172
#> NSE_y
#> 0.2257353In this case, the selected peaks algorithms correctly retain the biological variance and structural distances shown by high RV coefficient and Entropy Ratio.
Note on Base Peaks vs. Standard Mass Index: You
might notice that stdcluster$stdmassindex2 (Independent
Base Peaks) generally yields slightly better similarity scores than
std$stdmassindex (Independent Peaks). The original
GlobalStd algorithm purely uses PMD topology to filter redundancy
without prioritizing peak intensity. Consequently, it may select a minor
isotope or adduct peak as the representative feature, which is more
vulnerable to baseline noise. In contrast,
getpseudospectrum() intentionally selects the base
peak (the peak with the highest intensity) from each redundant PMD
cluster. Because base peaks have higher signal-to-noise ratios, they
capture the intrinsic biological variance of the underlying compound
more robustly, leading to representations that align more closely with
the uncompressed raw dataset.
Benchmark different datasets by Normalized Spectral Entropy (Sample Consistency Metric)
If you need to evaluate the internal dimensionality and information
purity of non-aligned peak tables (e.g., outputs spanning different
platforms or algorithms without same variables), use
getnse() to measure the Normalized Spectral Entropy.
# Calculate SCM/NSE for the full data and the reduced standard masses
getnse(std$data)
#> [1] 0.3641172
getnse(std$data[std$stdmassindex,])
#> [1] 0.3128792Higher values indicate greater intrinsic independence and lower redundancy clustering (noise padding) within the sample dimensions.
Structure/Reaction directed analysis
getsda function could be used to perform
Structure/reaction directed analysis. The cutoff of frequency is
automate found by PMD network analysis with the largest mean distance of
all nodes.
sda <- getsda(std)
#> PMD frequency cutoff is 6 by PMD network analysis with largest network average distance 6.67 .
#> 53 groups were found as high frequency PMD group.
#> 0 was found as high frequency PMD.
#> 1.98 was found as high frequency PMD.
#> 2.01 was found as high frequency PMD.
#> 2.02 was found as high frequency PMD.
#> 6.97 was found as high frequency PMD.
#> 11.96 was found as high frequency PMD.
#> 12 was found as high frequency PMD.
#> 13.98 was found as high frequency PMD.
#> 14.02 was found as high frequency PMD.
#> 14.05 was found as high frequency PMD.
#> 15.99 was found as high frequency PMD.
#> 16.03 was found as high frequency PMD.
#> 19.04 was found as high frequency PMD.
#> 28.03 was found as high frequency PMD.
#> 30.05 was found as high frequency PMD.
#> 31.99 was found as high frequency PMD.
#> 33.02 was found as high frequency PMD.
#> 37.02 was found as high frequency PMD.
#> 42.05 was found as high frequency PMD.
#> 48.04 was found as high frequency PMD.
#> 48.98 was found as high frequency PMD.
#> 49.02 was found as high frequency PMD.
#> 54.05 was found as high frequency PMD.
#> 56.06 was found as high frequency PMD.
#> 56.1 was found as high frequency PMD.
#> 58.04 was found as high frequency PMD.
#> 58.08 was found as high frequency PMD.
#> 58.11 was found as high frequency PMD.
#> 63.96 was found as high frequency PMD.
#> 66.05 was found as high frequency PMD.
#> 68.06 was found as high frequency PMD.
#> 70.04 was found as high frequency PMD.
#> 70.08 was found as high frequency PMD.
#> 74.02 was found as high frequency PMD.
#> 80.03 was found as high frequency PMD.
#> 82.08 was found as high frequency PMD.
#> 88.05 was found as high frequency PMD.
#> 91.1 was found as high frequency PMD.
#> 93.12 was found as high frequency PMD.
#> 94.1 was found as high frequency PMD.
#> 96.09 was found as high frequency PMD.
#> 101.05 was found as high frequency PMD.
#> 108.13 was found as high frequency PMD.
#> 110.11 was found as high frequency PMD.
#> 112.16 was found as high frequency PMD.
#> 116.08 was found as high frequency PMD.
#> 122.15 was found as high frequency PMD.
#> 124.16 was found as high frequency PMD.
#> 126.14 was found as high frequency PMD.
#> 144.18 was found as high frequency PMD.
#> 148.04 was found as high frequency PMD.
#> 150.2 was found as high frequency PMD.
#> 173.18 was found as high frequency PMD.Such largest mean distance of all nodes is calculated for top 1 to 100 (if possible) high frequency PMDs. Here is a demo for the network generation process.
library(igraph)
#>
#> Attaching package: 'igraph'
#> The following objects are masked from 'package:stats':
#>
#> decompose, spectrum
#> The following object is masked from 'package:base':
#>
#> union
cdf <- sda$sda
# get the PMDs and frequency
pmds <- as.numeric(names(sort(table(cdf$diff2),decreasing = T)))
freq <- sort(table(cdf$diff2),decreasing = T)
# filter the frequency larger than 10 for demo
pmds <- pmds[freq>10]
cdf <- sda$sda[sda$sda$diff2 %in% pmds,]
g <- igraph::graph_from_data_frame(cdf,directed = F)
l <- igraph::layout_with_fr(g)
for(i in 1:length(pmds)){
g2 <- igraph::delete_edges(g,which(E(g)$diff2%in%pmds[1:i]))
plot(g2,edge.width=1,vertex.label="",vertex.size=1,layout=l,main=paste('Top',length(pmds)-i,'high frequency PMDs'))
}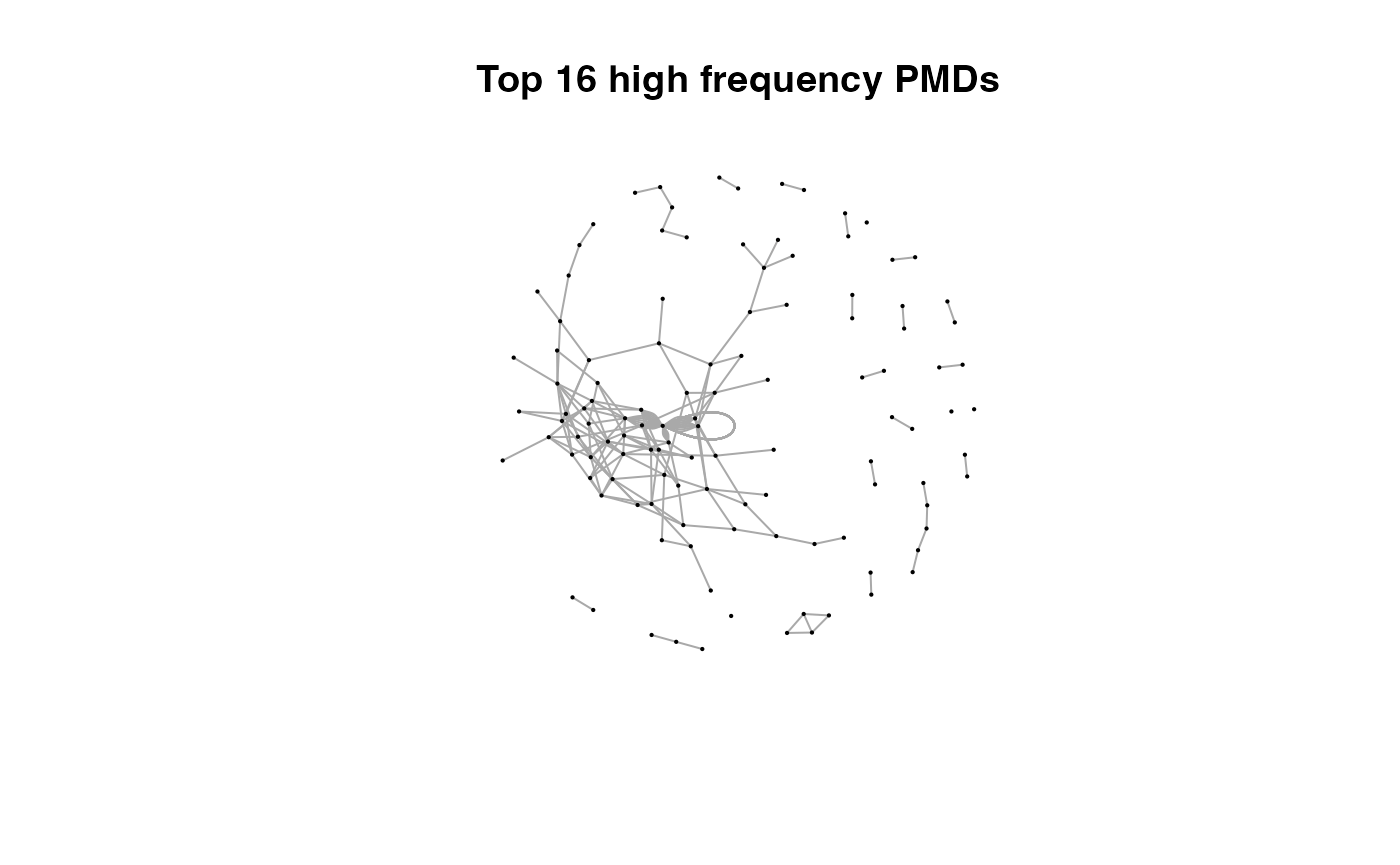
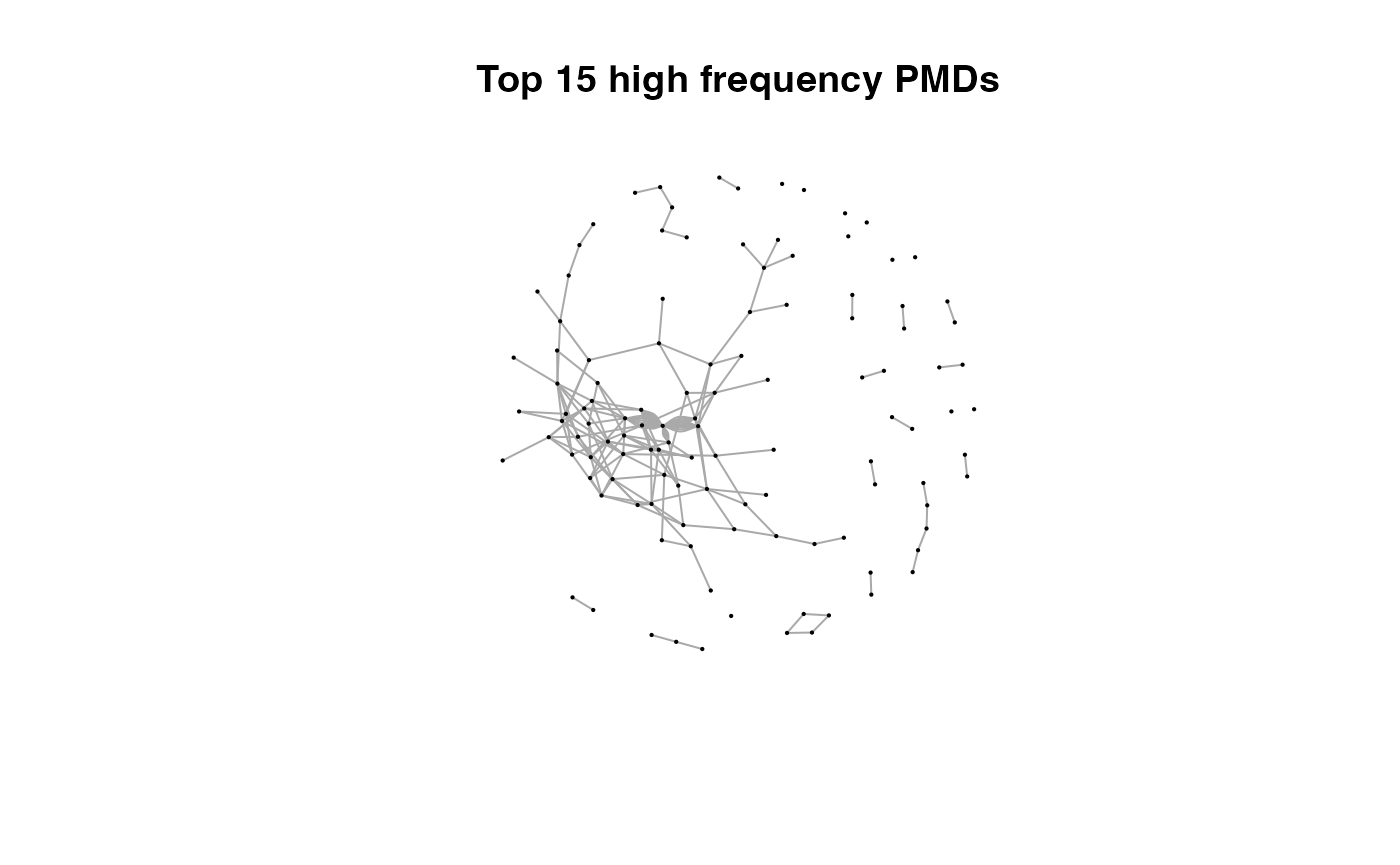
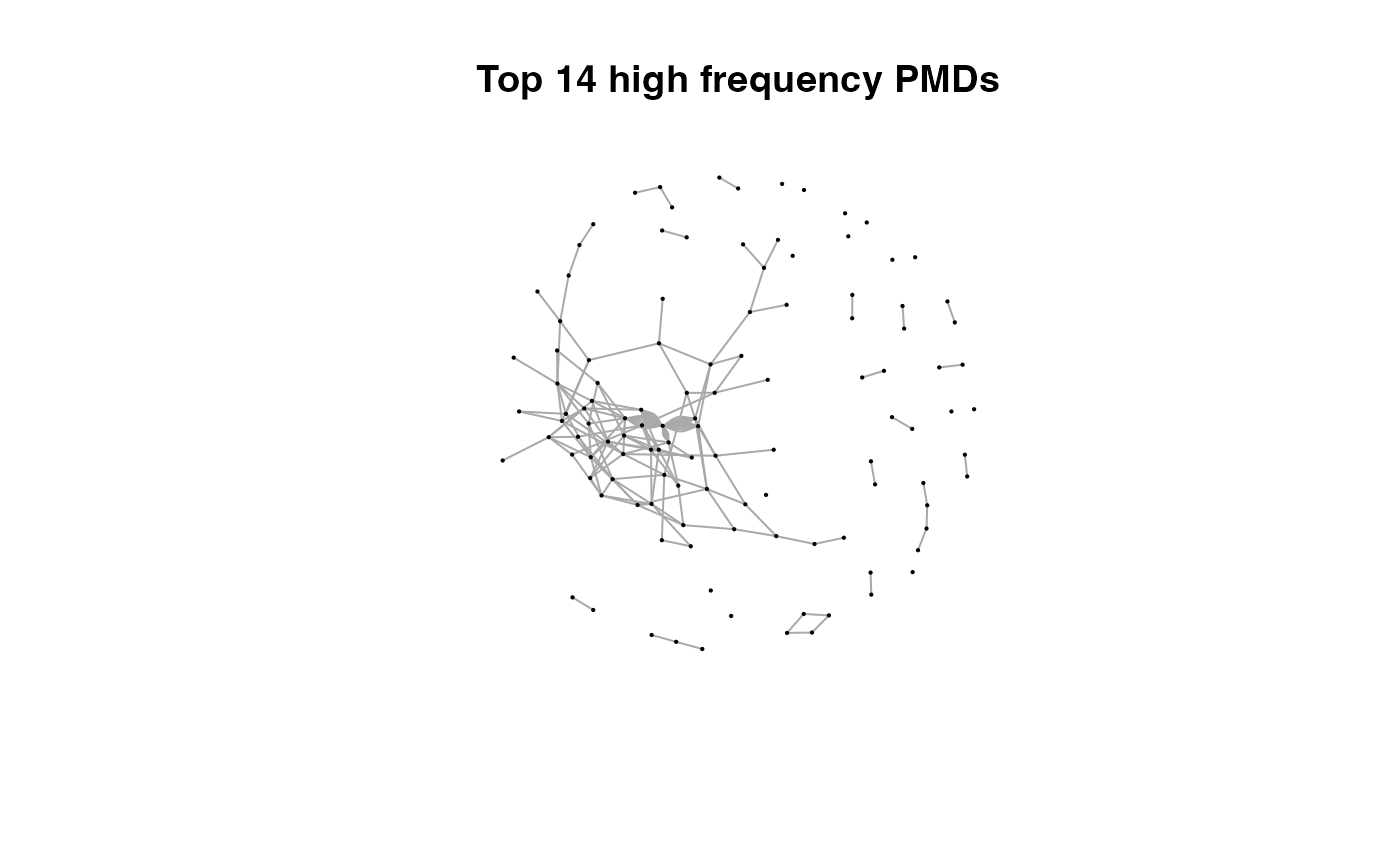
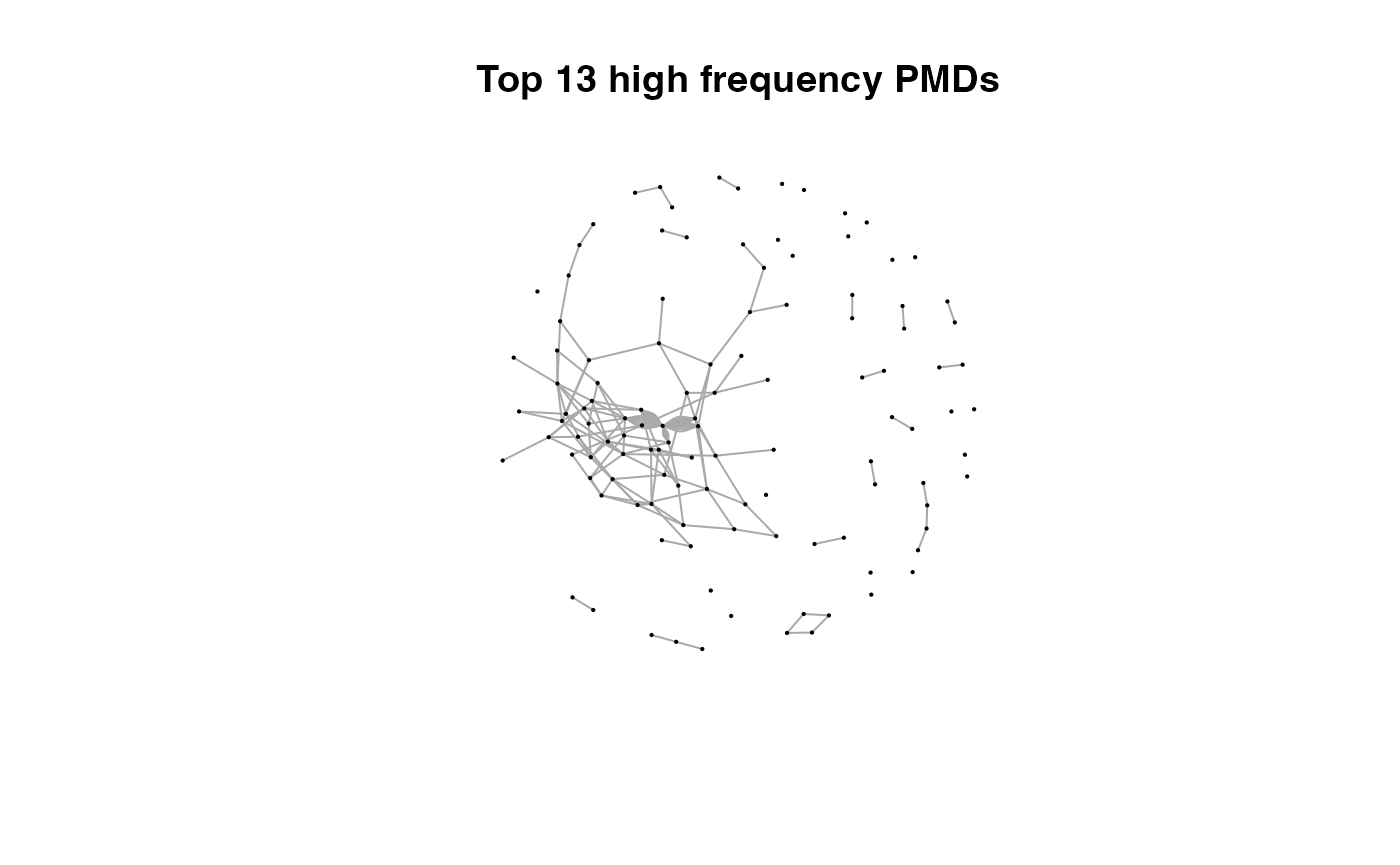
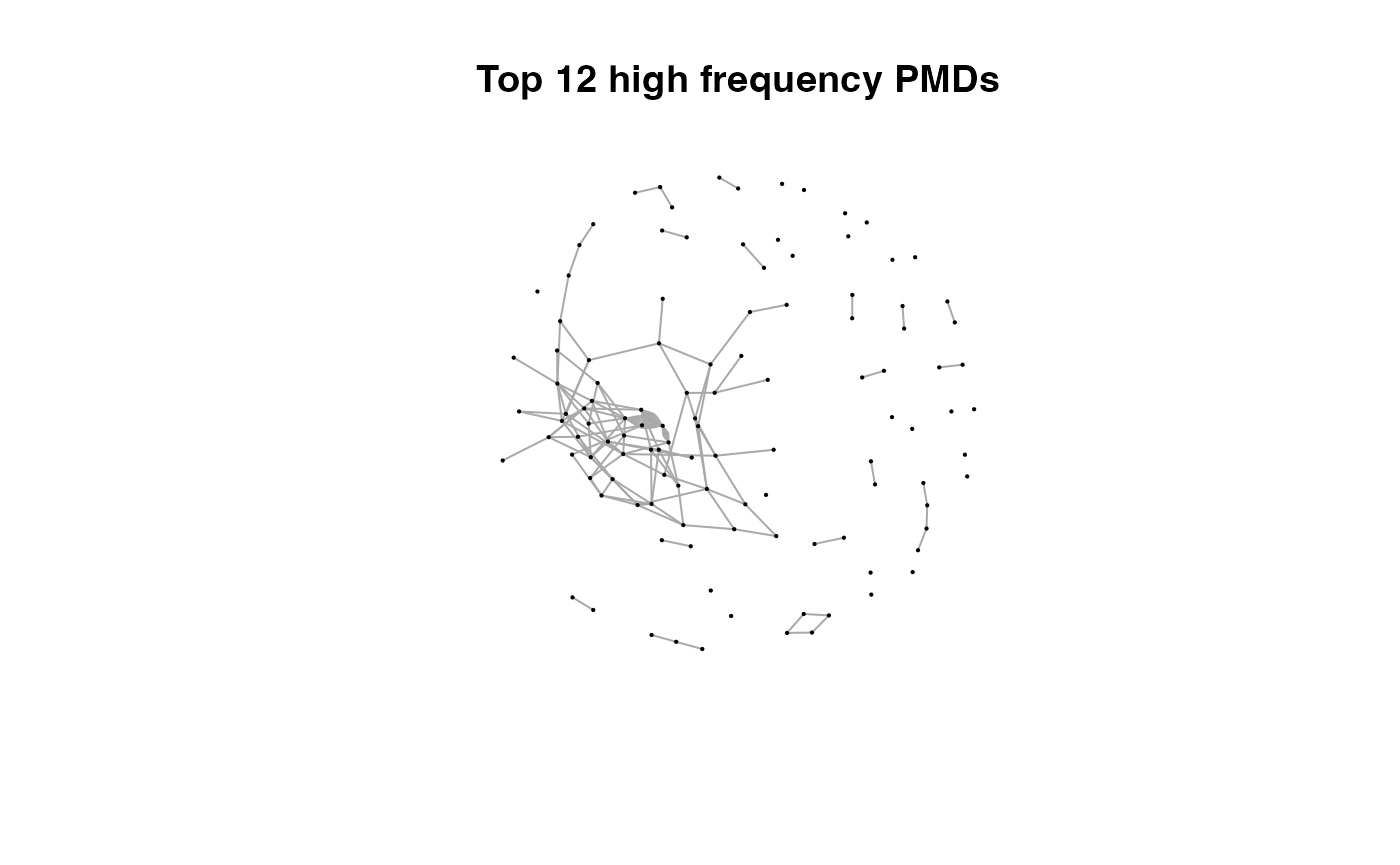
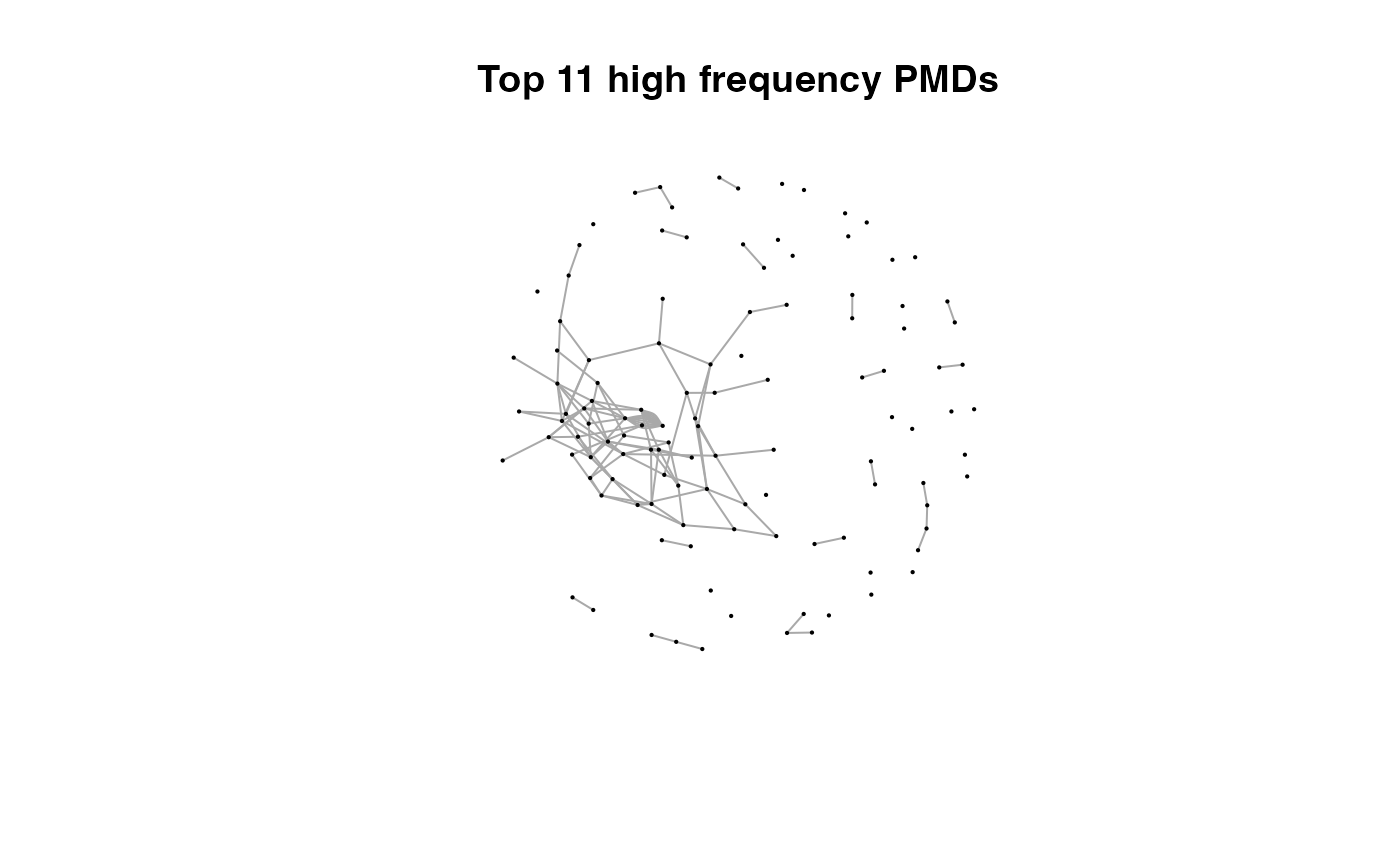
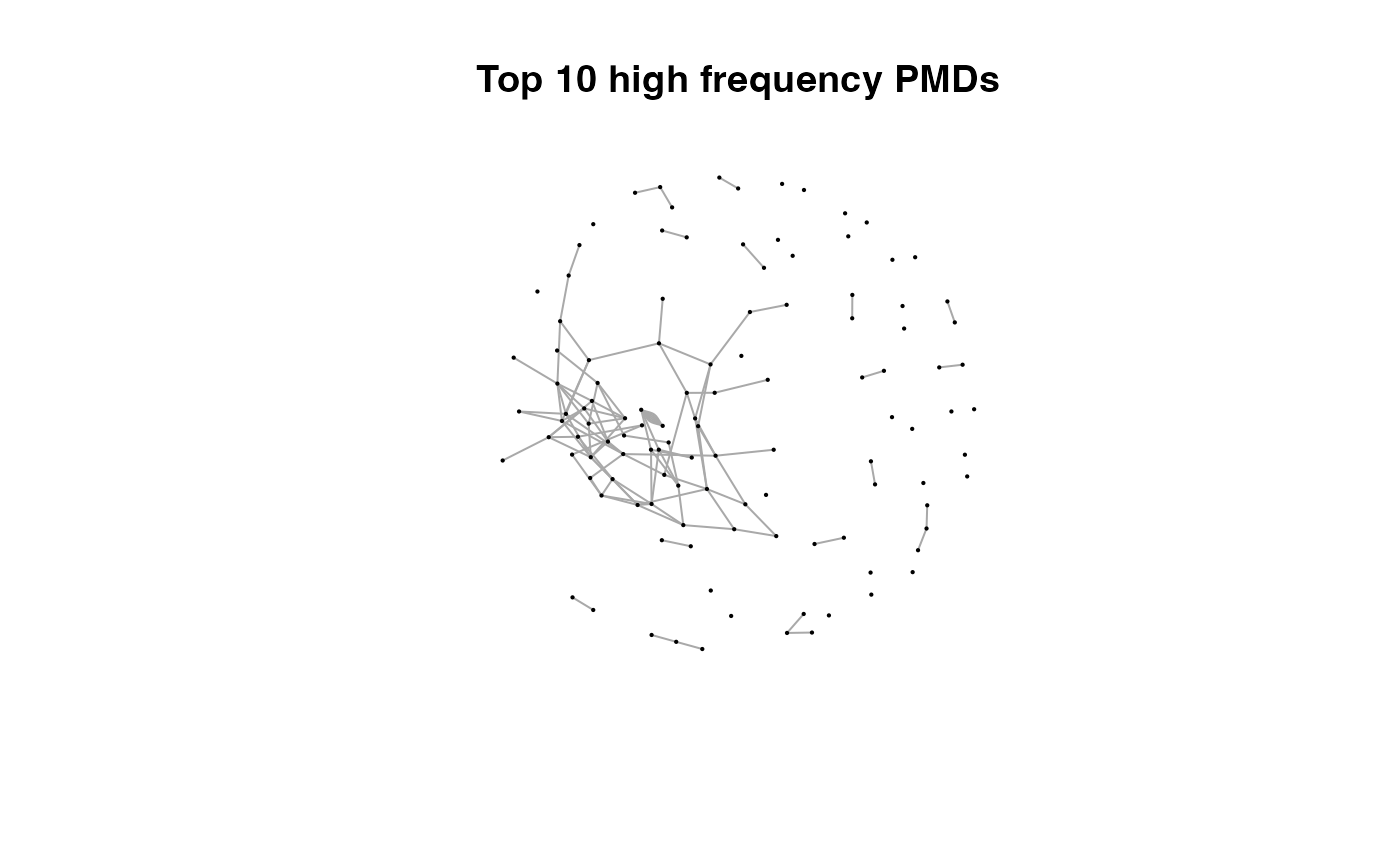
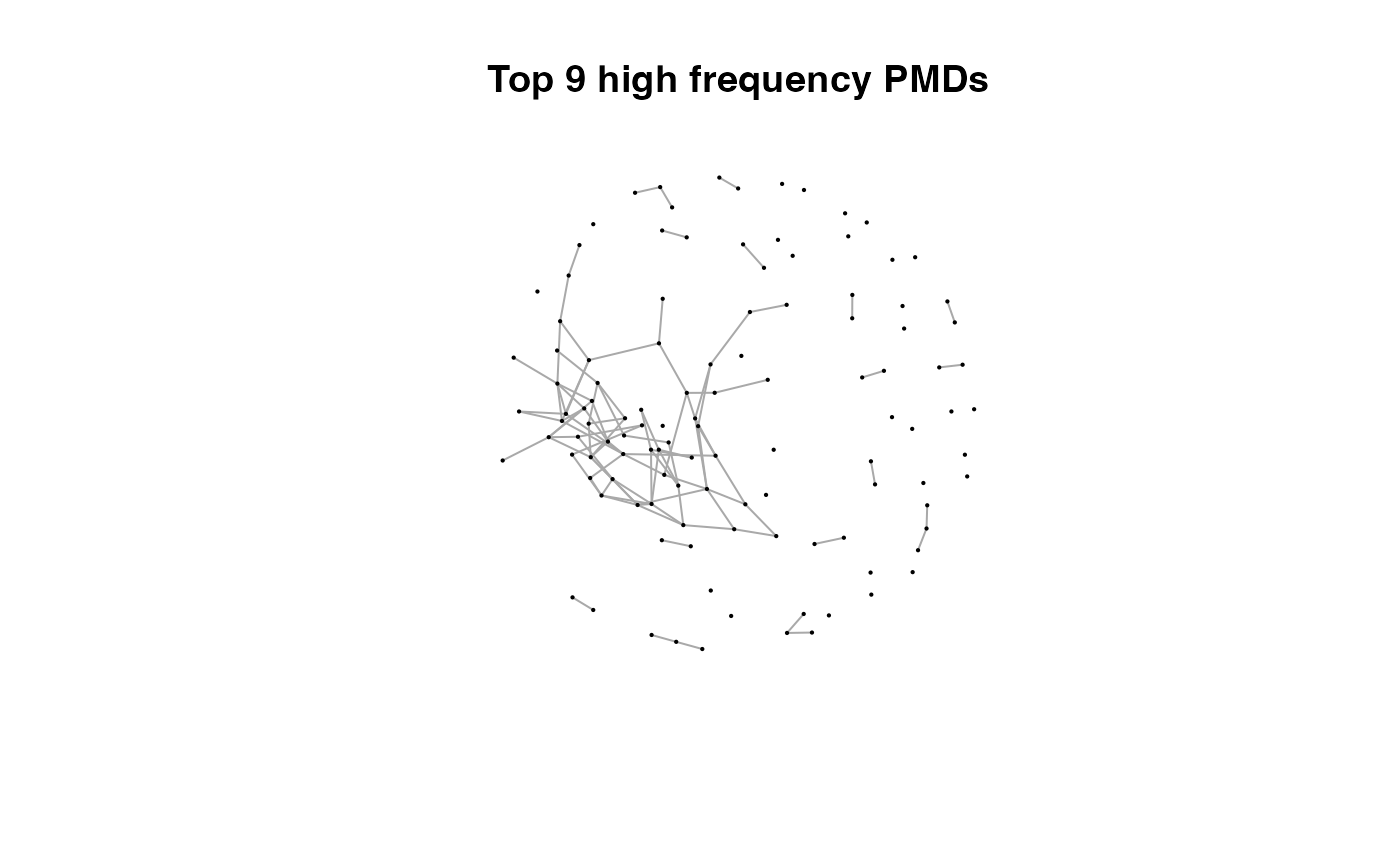
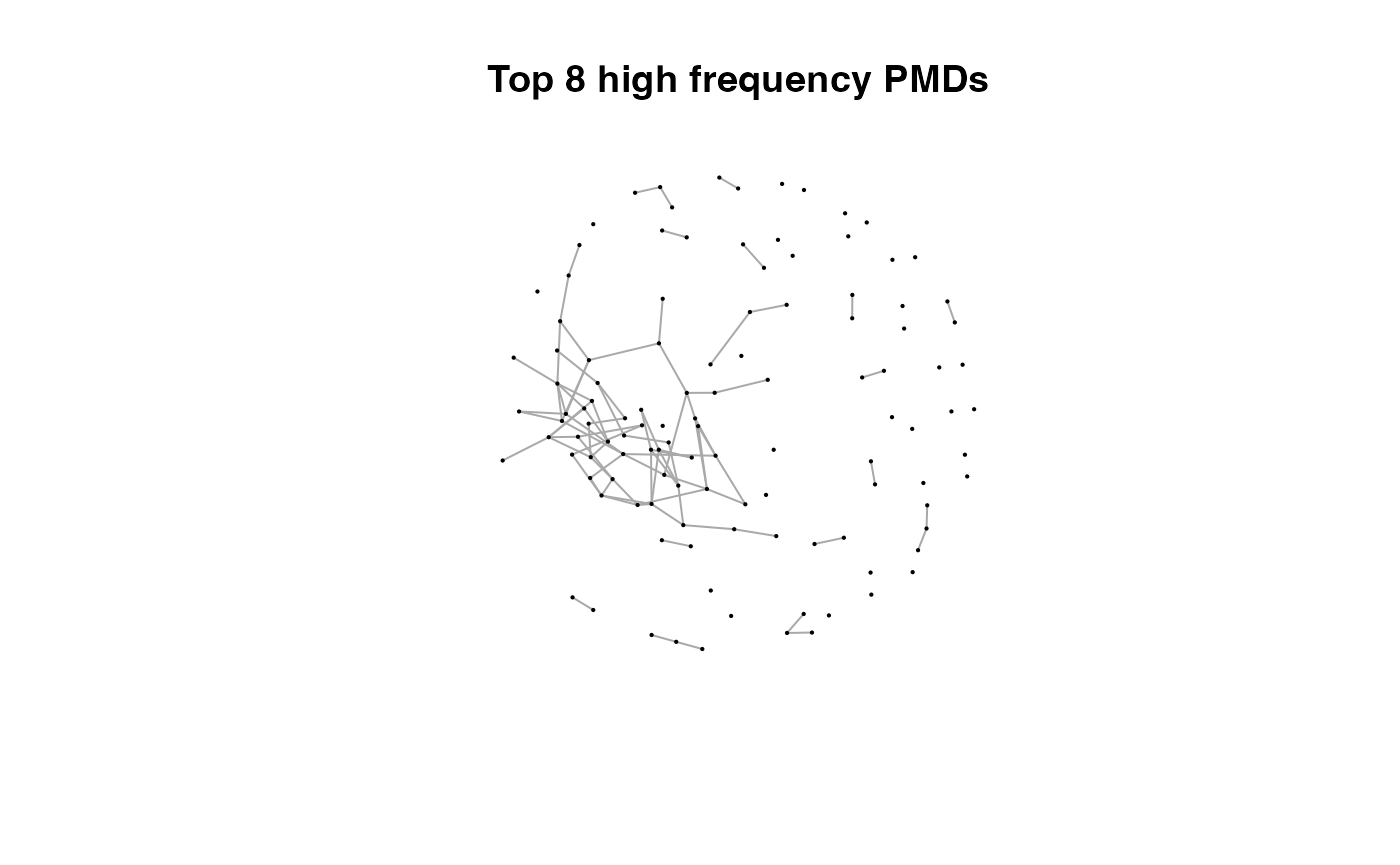
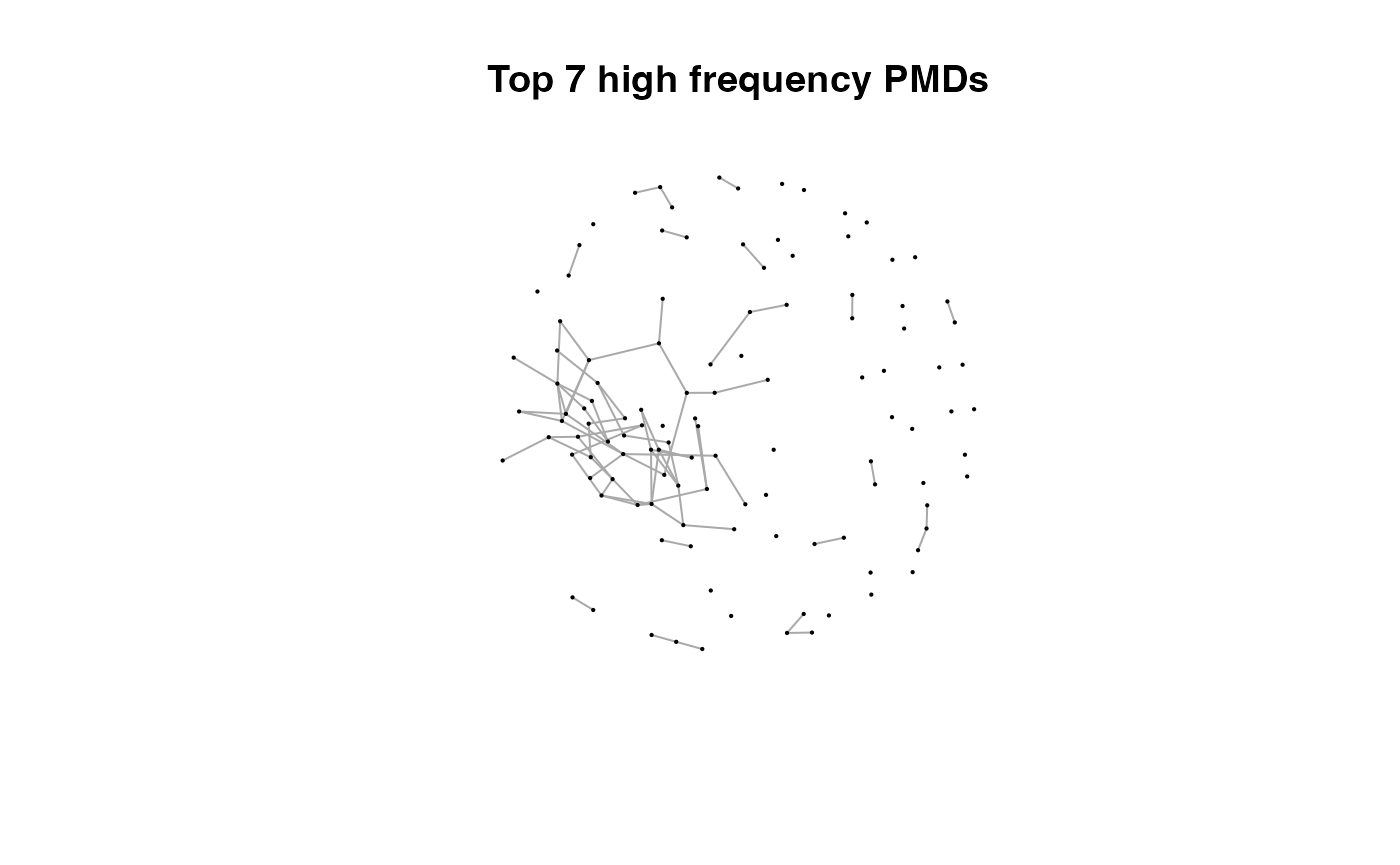
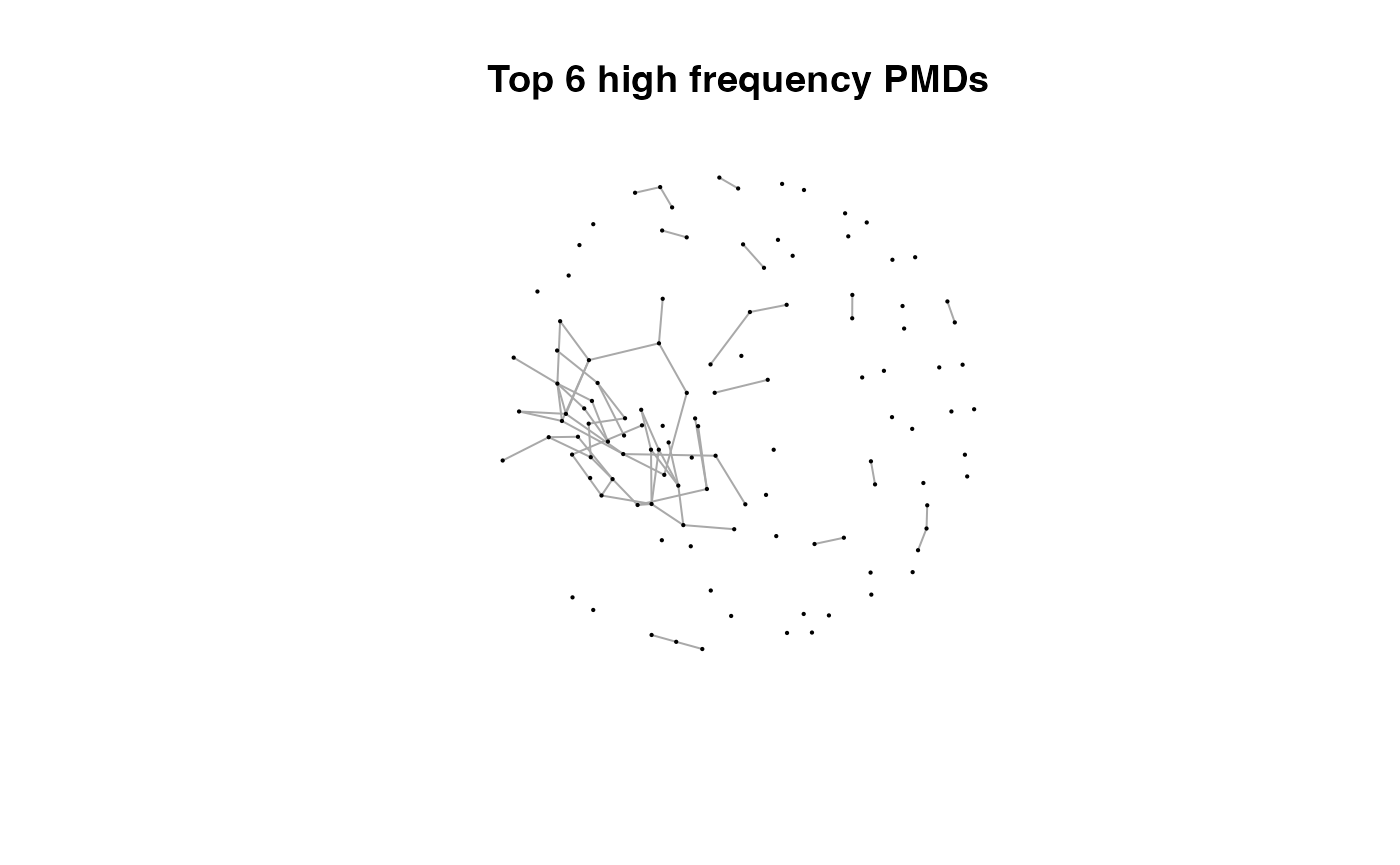


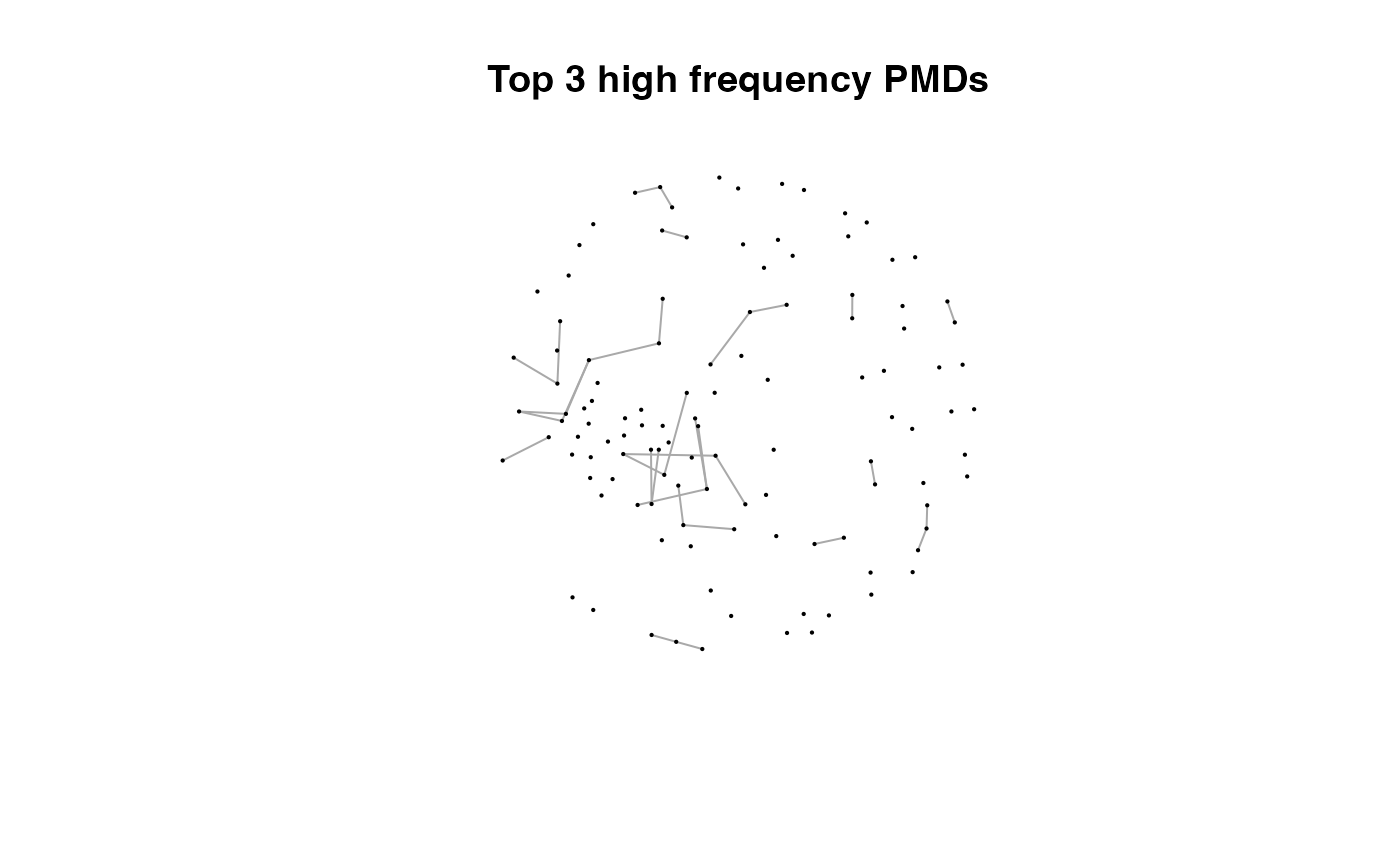


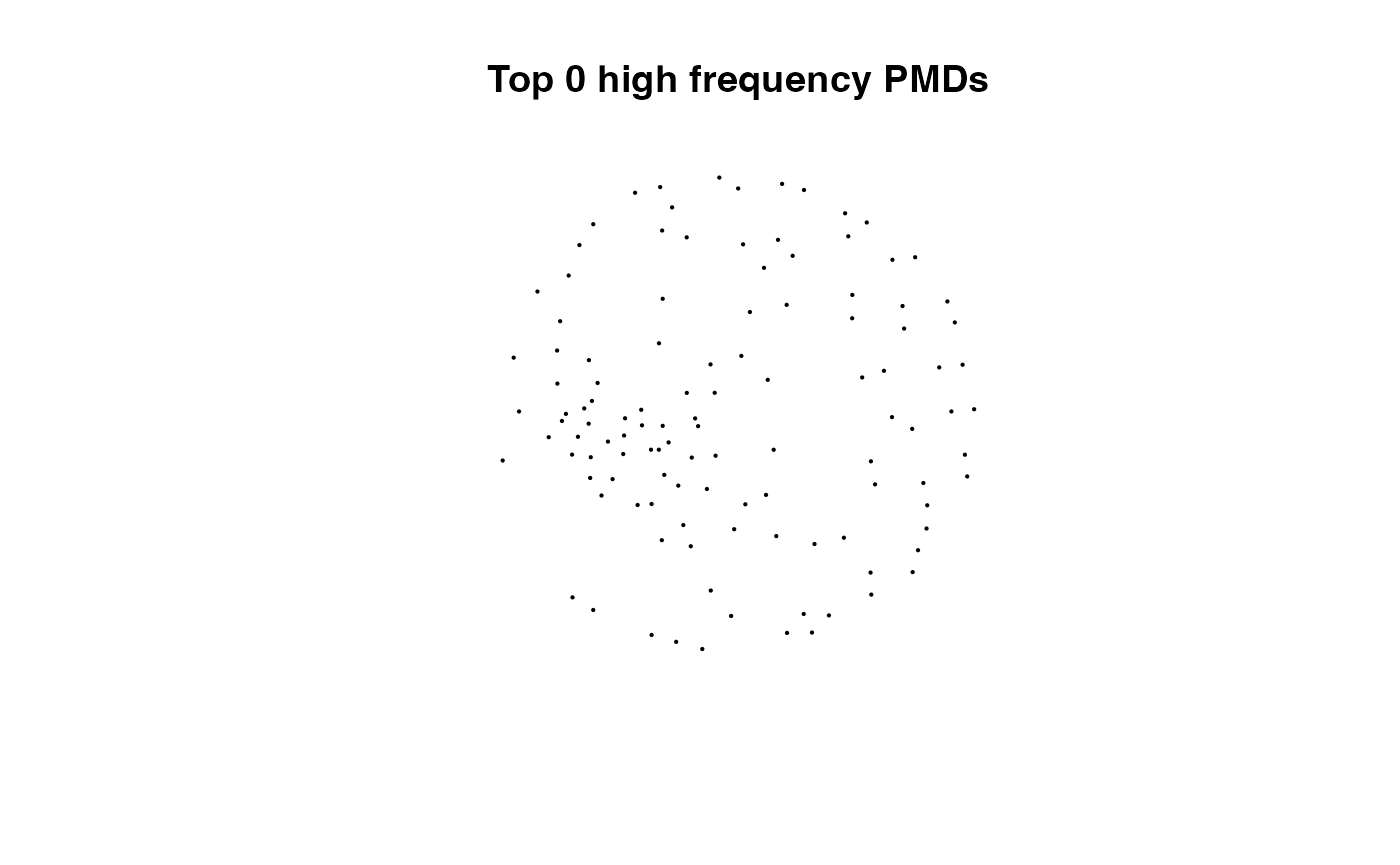
Here we could find more and more compounds will be connected with more high frequency PMDs. Meanwhile, the mean distance of all network nodes will increase. However, some PMDs are generated by random combination of ions. In this case, if we included those PMDs for the network, the mean distance of all network nodes will decrease. Here, the largest mean distance means no more information will be found for certain data set and such value is used as the cutoff for high frequency PMDs selection.
You could use plotstdsda to show the distribution of the
selected paired peaks.
plotstdsda(sda)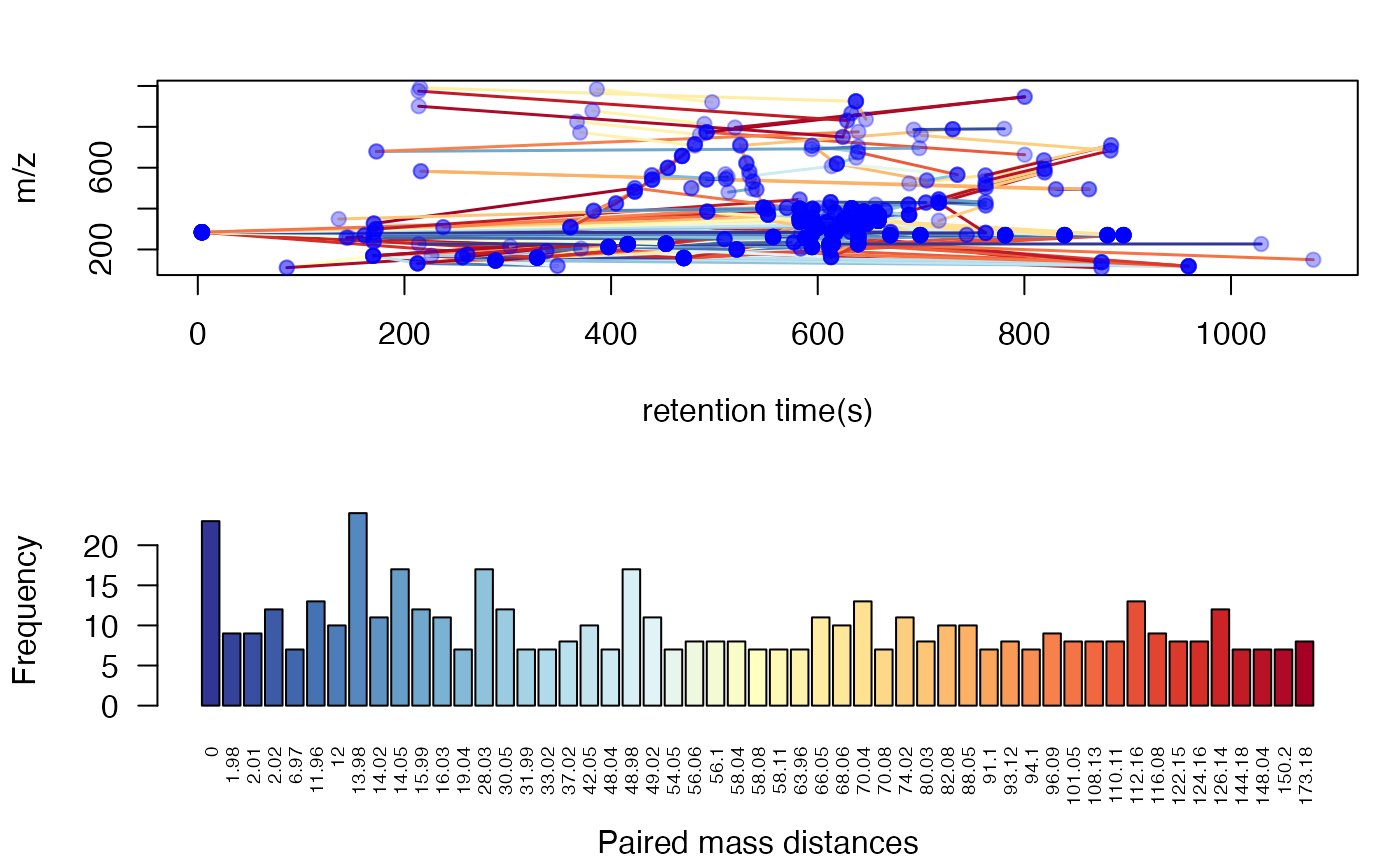
You could also use index to show the distribution of certain PMDs.
par(mfrow = c(1,3),mar = c(4,4,2,1)+0.1)
plotstdsda(sda,sda$sda$diff2 == 2.02)
plotstdsda(sda,sda$sda$diff2 == 28.03)
plotstdsda(sda,sda$sda$diff2 == 58.04)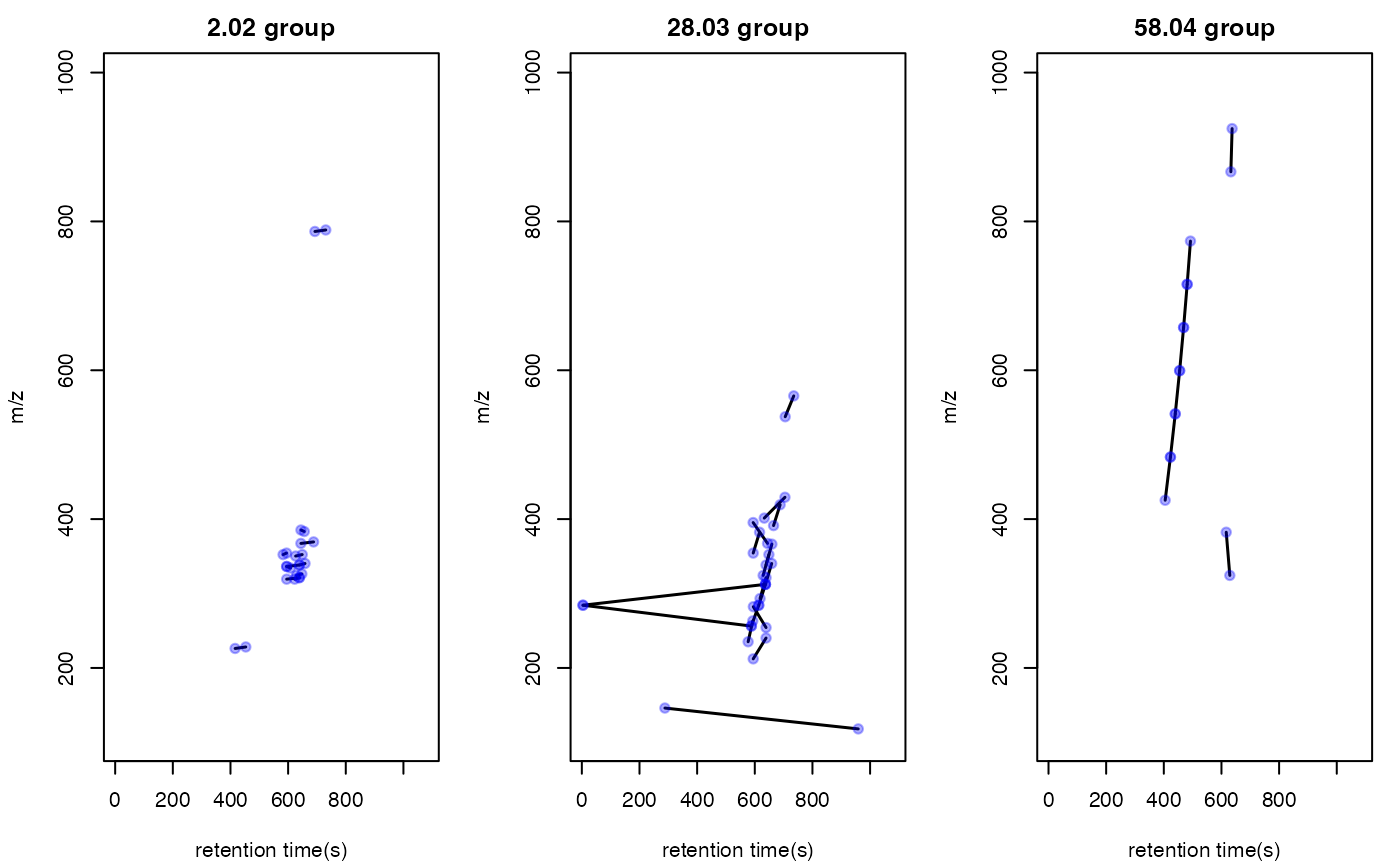
Structure/reaction directed analysis could be directly performed on all the peaks, which is slow to process:
sdaall <- getsda(spmeinvivo)
#> PMD frequency cutoff is 104 by PMD network analysis with largest network average distance 14.06 .
#> 6 groups were found as high frequency PMD group.
#> 0 was found as high frequency PMD.
#> 2.02 was found as high frequency PMD.
#> 28.03 was found as high frequency PMD.
#> 31.01 was found as high frequency PMD.
#> 58.04 was found as high frequency PMD.
#> 116.08 was found as high frequency PMD.
par(mfrow = c(1,3),mar = c(4,4,2,1)+0.1)
plotstdsda(sdaall,sdaall$sda$diff2 == 2.02)
plotstdsda(sdaall,sdaall$sda$diff2 == 28.03)
plotstdsda(sdaall,sdaall$sda$diff2 == 58.04)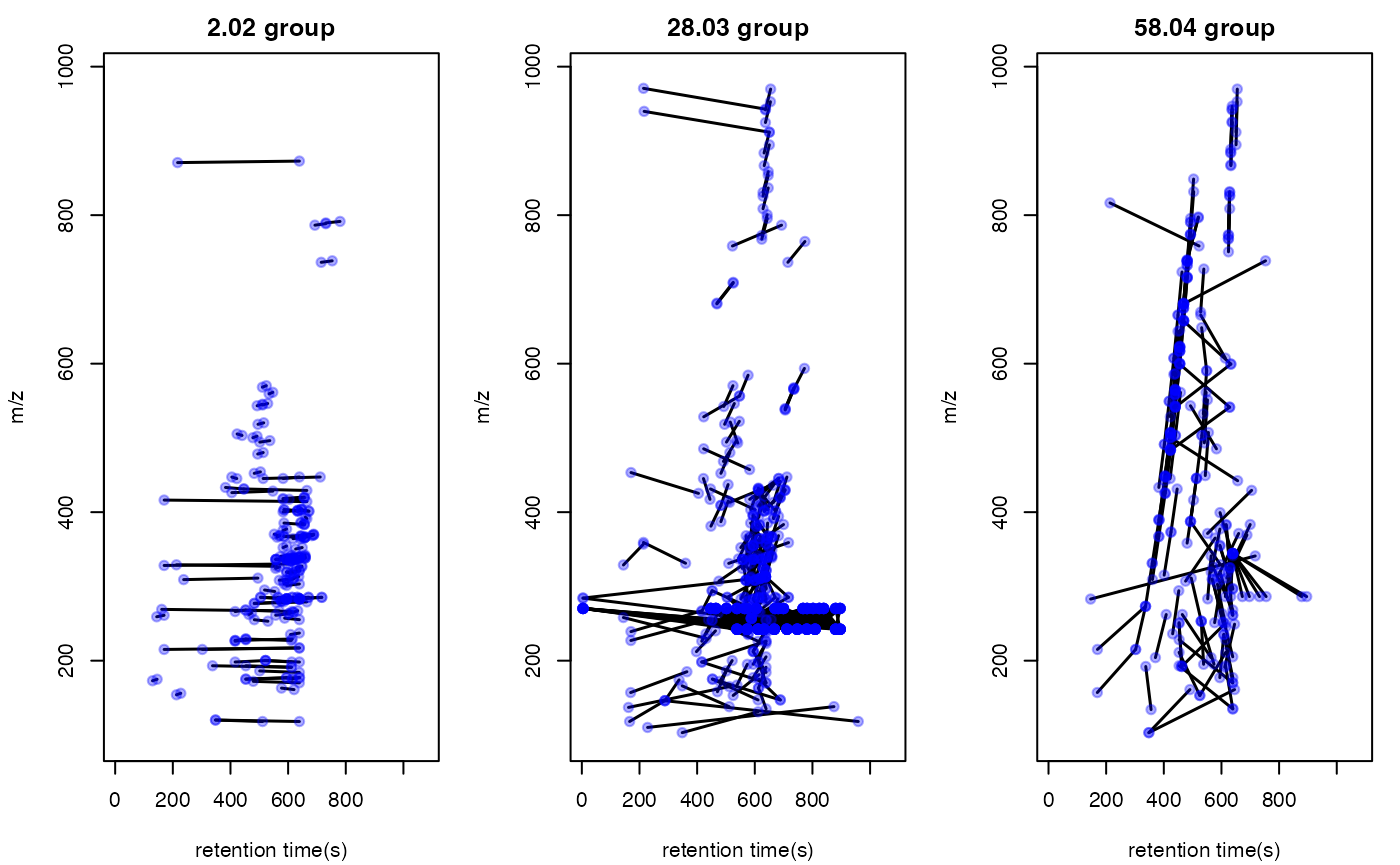
Extra filter with correlation coefficient cutoff
Structure/Reaction directed analysis could also use correlation to restrict the paired ions. However, similar to GlobalStd algorithm, such cutoff will remove low intensity data. Researcher should have a clear idea to use this cutoff.
sda2 <- getsda(std, corcutoff = 0.9)
#> PMD frequency cutoff is 6 by PMD network analysis with largest network average distance 6.67 .
#> 41 groups were found as high frequency PMD group.
#> 0 was found as high frequency PMD.
#> 1.98 was found as high frequency PMD.
#> 2.01 was found as high frequency PMD.
#> 2.02 was found as high frequency PMD.
#> 11.96 was found as high frequency PMD.
#> 12 was found as high frequency PMD.
#> 13.98 was found as high frequency PMD.
#> 14.02 was found as high frequency PMD.
#> 14.05 was found as high frequency PMD.
#> 15.99 was found as high frequency PMD.
#> 16.03 was found as high frequency PMD.
#> 19.04 was found as high frequency PMD.
#> 28.03 was found as high frequency PMD.
#> 30.05 was found as high frequency PMD.
#> 31.99 was found as high frequency PMD.
#> 33.02 was found as high frequency PMD.
#> 42.05 was found as high frequency PMD.
#> 48.98 was found as high frequency PMD.
#> 49.02 was found as high frequency PMD.
#> 54.05 was found as high frequency PMD.
#> 56.06 was found as high frequency PMD.
#> 58.04 was found as high frequency PMD.
#> 58.08 was found as high frequency PMD.
#> 63.96 was found as high frequency PMD.
#> 66.05 was found as high frequency PMD.
#> 68.06 was found as high frequency PMD.
#> 70.08 was found as high frequency PMD.
#> 74.02 was found as high frequency PMD.
#> 80.03 was found as high frequency PMD.
#> 82.08 was found as high frequency PMD.
#> 88.05 was found as high frequency PMD.
#> 93.12 was found as high frequency PMD.
#> 94.1 was found as high frequency PMD.
#> 96.09 was found as high frequency PMD.
#> 108.13 was found as high frequency PMD.
#> 110.11 was found as high frequency PMD.
#> 112.16 was found as high frequency PMD.
#> 116.08 was found as high frequency PMD.
#> 122.15 was found as high frequency PMD.
#> 124.16 was found as high frequency PMD.
#> 126.14 was found as high frequency PMD.
plotstdsda(sda2)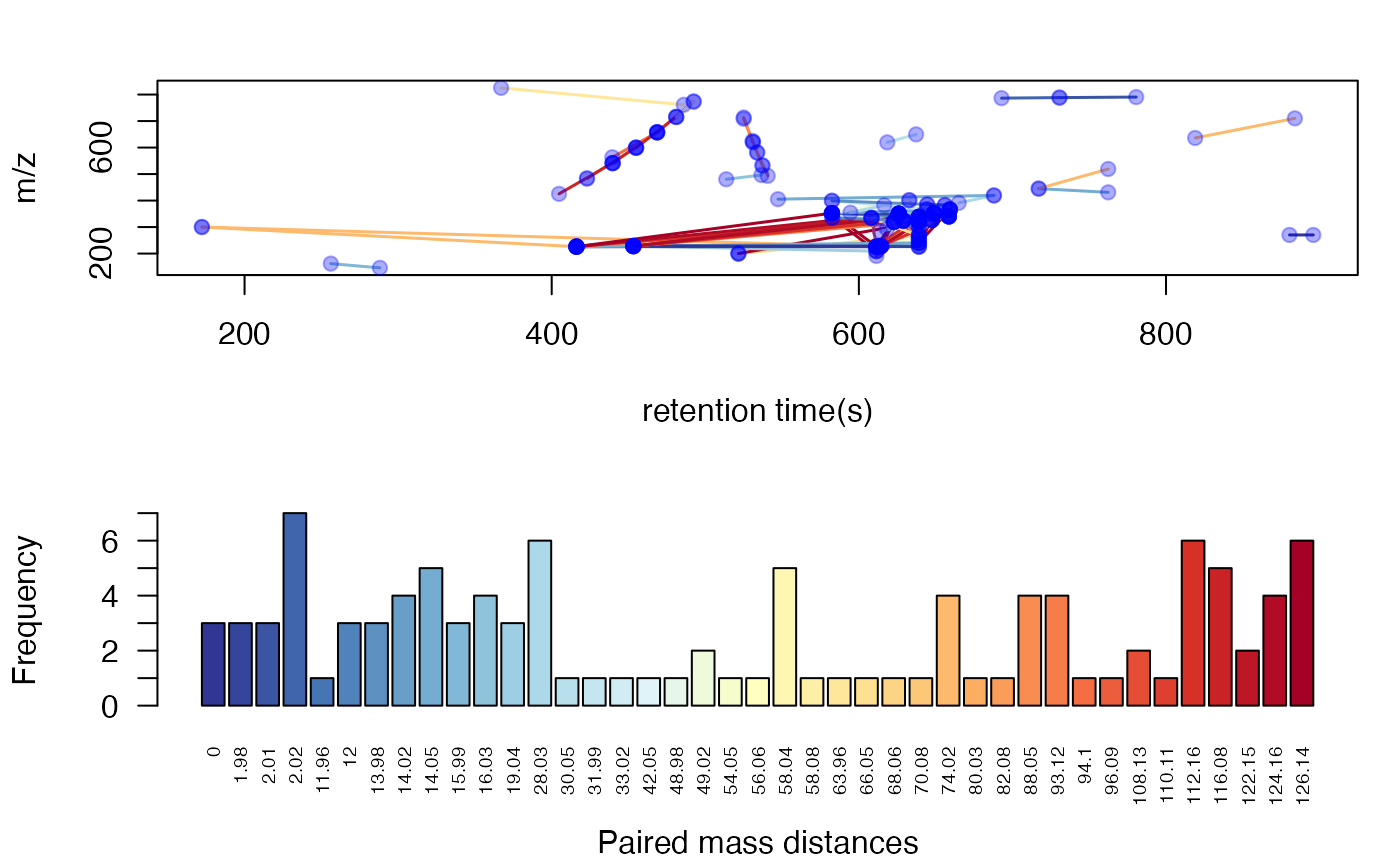
Structure/reaction directed analysis for peaks/compounds only data
When you only have data of peaks without retention time or compounds
list, structure/reaction directed analysis could also be done by
getrda function.
sda <- getrda(spmeinvivo$mz)
#> 164462 pmd found.
#> 20 pmd used.
# check high frequency pmd
colnames(sda)
#> [1] "0" "1.001" "1.002" "1.003" "1.004" "2.015" "2.016"
#> [8] "14.015" "17.026" "18.011" "21.982" "28.031" "28.032" "44.026"
#> [15] "67.987" "67.988" "88.052" "116.192" "135.974" "135.975"
# get certain pmd related m/z
idx <- sda[,'2.016']
# show the m/z
spmeinvivo$mz[idx]
#> [1] 118.0651 118.0652 120.0812 159.1575 162.0552 170.0330 170.0932 170.1541
#> [9] 174.1363 174.9917 175.0873 176.0305 176.0418 181.9872 184.1695 188.6484
#> [17] 192.1487 192.1604 226.9522 226.9523 228.1969 228.1973 259.1148 261.1317
#> [25] 270.3185 271.3217 272.3230 272.3234 273.8902 274.8744 284.2955 285.3002
#> [33] 285.3002 286.3101 286.3101 291.0712 293.1755 294.9392 296.2961 304.3081
#> [41] 305.2480 305.3118 308.0889 308.2953 308.2954 309.1672 309.2046 315.1781
#> [49] 317.9344 319.3005 319.3002 319.9302 320.3041 320.3322 321.3165 322.3185
#> [57] 323.3221 324.3266 325.3294 327.2022 327.3449 329.0052 331.0031 350.3426
#> [65] 352.3214 352.3215 353.3244 354.3365 355.0696 359.2410 361.2353 372.3197
#> [73] 375.3066 383.2804 383.3723 384.3350 385.2753 385.3480 387.2851 397.1907
#> [81] 399.3274 400.9174 401.3420 403.2859 432.8860 433.2781 445.8289 447.1173
#> [89] 451.3633 462.8615 522.3557 524.1178 525.9831 526.4841 705.7223 708.8218
#> [97] 976.3139 976.8122 982.7763Wrap function for GlobalStd algorithm
globalstd function is a wrap function to process
GlobalStd algorithm and structure/reaction directed analysis in one
line. All the plot function could be directly used on the
list objects from globalstd function. If you
want to perform structure/reaction directed analysis, set the
sda=T in the globalstd function.
result <- globalstd(spmeinvivo, sda=FALSE)
#> 75 retention time clusters found.
#> Using ng = 15
#> 5 unique PMDs retained.
#> The unique within RT clusters high frequency PMD(s) is(are) 28.03 21.98 44.03 17.03 18.01.
#> 409 isotope peaks found.
#> 109 multiple charged isotope peaks found.
#> 4 multiple charged peaks found.
#> 346 paired peaks found.
#> 8 group(s) have single peaks 14 23 32 33 54 55 56 75
#> 11 group(s) with multiple peaks while no isotope/paired relationship 4 5 7 8 11 ... 42 49 68 72 73
#> 9 group(s) with isotope without paired relationship 2 9 22 26 52 62 64 66 70
#> 4 group(s) with paired without isotope relationship 1 10 15 18
#> 43 group(s) with both paired and isotope relationship 3 6 12 13 16 ... 65 67 69 71 74
#> 291 standard masses identified.Use independent peaks for MS/MS validation (PMDDA)
Independent peaks are supposing generated from different compounds.
We could use those peaks for MS/MS analysis instead of DIA or DDA. Here
we need multiple injections for one sample since it might be impossible
to get all ions’ fragment ions in one injection with good sensitivity.
You could use gettarget to generate the index for the
injections and output the peaks for each run.
# you need retention time for independent peaks
index <- gettarget(std$rt[std$stdmassindex])
#> You need 10 injections!
# output the ions for each injection
table(index)
#> index
#> 1 2 3 4 5 6 7 8 9 10
#> 25 28 40 18 28 29 34 45 28 16
# show the ions for the first injection
std$mz[index==1]
#> [1] 110.0717 114.0918 122.9247 125.9874 133.0794 137.9879 141.9594 149.9530
#> [9] 165.0787 166.0867 172.1705 174.9383 175.1482 183.0809 193.1599 198.1854
#> [17] 203.0532 205.0872 205.1954 220.1184 226.1823 242.2863 242.9260 255.2511
#> [25] 259.9651 264.1944 267.0639 270.3185 270.3172 270.3185 270.3184 271.3216
#> [33] 273.8902 285.3002 286.3101 294.2054 296.9066 300.2046 306.2514 310.3117
#> [41] 315.1781 317.9344 319.3005 319.9302 329.8840 334.3101 349.3476 351.3455
#> [49] 363.3114 367.2694 371.3345 374.3041 380.0033 382.3673 383.2804 386.3523
#> [57] 391.2835 394.8754 410.2585 411.0941 417.2462 420.3193 424.0815 430.0895
#> [65] 437.2355 445.2767 445.3874 446.8878 447.2935 467.1031 471.3317 505.1055
#> [73] 507.3303 527.2976 536.1655 540.8890 542.3991 548.2764 551.3562 556.8633
#> [81] 560.3877 564.1885 567.1783 586.4524 589.5800 600.4401 605.2231 608.4285
#> [89] 620.4358 636.1983 643.4632 646.4585 649.6613 650.8519 669.4185 675.5084
#> [97] 703.3651 704.1390 720.8364 732.5472 739.6479 750.6089 758.4735 760.8210
#> [105] 764.8339 771.8544 773.6523 779.5153 803.5434 804.8442 816.5102 826.6806
#> [113] 832.8212 840.8392 861.5014 870.7857 872.3052 872.8314 873.3343 889.8086
#> [121] 900.3092 942.7638 946.7164 979.7901 991.7894
std$rt[index==1]
#> [1] 228.0840 172.8550 217.9690 1079.6500 212.6560 165.4680 1079.4300
#> [8] 1079.6400 511.3690 348.1340 478.9360 216.1500 453.1780 533.7950
#> [15] 453.1780 415.9985 144.8840 583.5530 639.1010 170.8240 611.4120
#> [22] 780.5760 216.0600 639.1000 145.9660 473.1445 568.7680 538.0815
#> [29] 4.0610 604.7690 465.0070 781.0040 145.9680 716.7800 631.8770
#> [36] 452.0005 145.1090 172.2230 599.6270 618.4820 401.2135 145.5170
#> [43] 622.7690 145.2280 144.3380 608.1990 659.2440 626.0920 561.2685
#> [50] 382.6770 551.7325 582.4840 212.9175 616.5550 547.0180 644.4580
#> [57] 665.0280 217.1550 633.9130 563.1970 444.6075 687.8085 583.9830
#> [64] 717.1010 170.9435 421.8915 582.4825 215.6320 404.5340 717.1030
#> [71] 540.8660 762.4675 554.8390 493.5080 762.5770 213.7260 439.6780
#> [78] 493.5080 551.4100 215.9830 533.3660 762.5770 762.3630 439.2500
#> [85] 889.0520 455.1500 762.5750 613.3395 531.0080 818.9800 449.3200
#> [92] 605.8410 637.2775 215.6410 527.7950 468.2215 213.5480 639.1000
#> [99] 215.1360 481.0785 639.1005 624.2710 522.4370 215.6320 213.7270
#> [106] 213.7720 613.5550 519.6690 665.0290 213.5090 213.3340 628.4480
#> [113] 213.7130 213.9270 213.3395 216.5120 639.0990 213.5090 213.5480
#> [120] 215.2605 213.5480 636.9060 637.1710 213.9410 215.3500Shiny application
An interactive document has been included in this package to perform
PMD analysis. You need to prepare a csv file with m/z and retention time
of peaks. Such csv file could be generated by run
enviGCMS::getcsv() on the list object from
enviGCMS::getmzrt(xset) function. The xset
should be XCMSnExp object or xcmsSet object.
You could also generate the csv file by
enviGCMS::getmzrt(xset,name = 'test'). You will find the
csv file in the working dictionary named test.csv.
Then you could run runPMD() to start the Graphical user
interface(GUI) for GlobalStd algorithm and structure/reaction directed
analysis.